Please use the new website www.giottosuite.com. This website is outdated and archived for consistency with the original Giotto publication (Dries et al, Genome Biology, 2021).
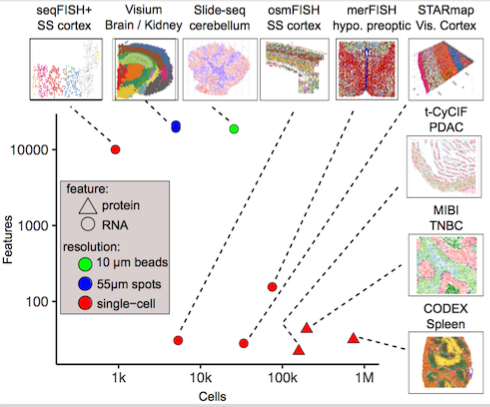
The Giotto package consists of two modules, Giotto Analyzer and Viewer (see www.spatialgiotto.com), which provide tools to process, analyze and visualize single-cell spatial expression data. The underlying framework is generalizable to virtually all currently available spatial datasets. We recently demonstrated the general applicability on 10 different datasets created by 9 different state-of-the-art spatial technologies, including in situ hybridization (seqFISH+, merFISH, osmFISH), sequencing (Slide-seq, Visium, STARmap) and imaging-based multiplexing/proteomics (CyCIF, MIBI, CODEX). These technologies differ in terms of resolution (single cell vs multiple cells), spatial dimension (2D vs 3D), molecular modality (protein vs RNA), and throughput (number of cells and genes). More information and documentation about the latest released version of Giotto Analyzer can be found at https://rubd.github.io/Giotto_site/.
Requirements
- R (>= 3.5.1)
- Python (>= 3.0)
- Windows, MacOS or Linux specific installation tools. See link.
Installation
See FAQs for additional information.
Required python modules
These are necessary to run all available analyses, but can be installed automatically.
Required python modules:
- pandas
- python-igraph (igraph)
- networkx
- leidenalg
- python-louvain (community)
- smfishHmrf
- python.app (!!OSX only!!)
- scikit-learn
Automatic installation
The python modules will be installed automatically in a miniconda environment when installing Giotto. However, it will ask you whether you want to install them and you can opt out and select your preferred python path. In that case you need to do a manual installation of the python modules.
Examples
- see https://github.com/RubD/spatial-datasets to find raw and pre-processed input data and Giotto scripts (in progress).
- typical run time range for the different datasets on a personal computer is around 10~45 mins.
- click on the image and try them out yourself.
- all examples are gradually updated to the latest Giotto version [work in progress]
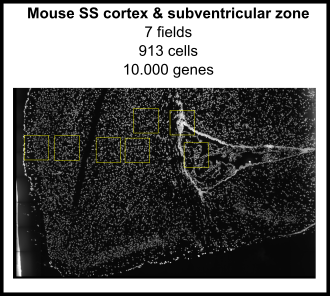
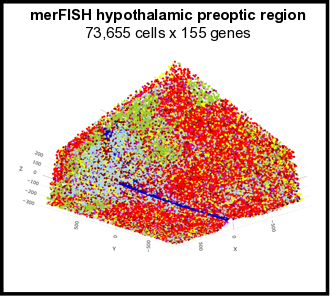
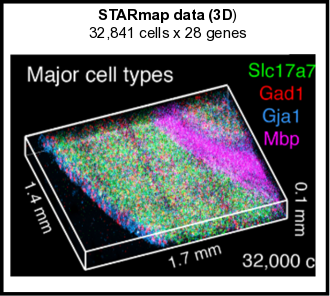
References
Dries, R., Zhu, Q. et al. Giotto, a toolbox for integrative analysis and visualization of spatial expression data. bioRxiv 701680 (2019). doi:10.1101/701680
Eng, C.-H. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 1 (2019). doi:10.1038/s41586-019-1049-y
Zhu, Q., Shah, S., Dries, R., Cai, L. & Yuan, G.-C. Identification of spatially associated subpopulations by combining scRNAseq and sequential fluorescence in situ hybridization data. Nature Biotechnology (2018). doi:10.1038/nbt.4260