Source:
vignettes/getting_started.Rmd
getting_started.RmdDescription
- Giotto provides a flexible framework for common single-cell
processing steps such as:
- quality control
- normalization
- dimension reduction
- clustering and cell type annotation
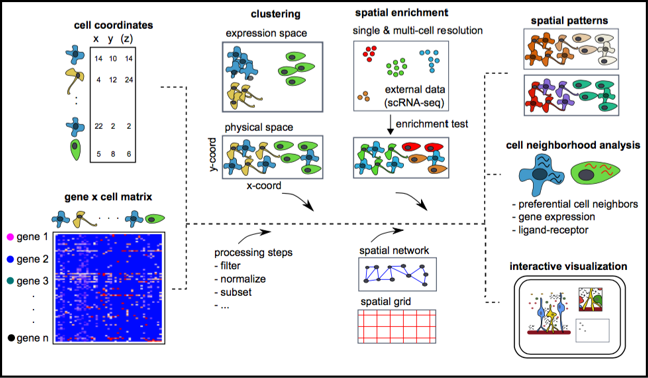
- To facilitate the analysis of recently emerging high-throughput, but lower-resolution spatial transcriptomic technologies, such as 10X Genomics Visium and Slide-seq, Giotto has 3 implemented algorithms for estimating the spatial enrichment of different cell types by integration of known gene signatures or single-cell RNAseq expression and annotation data.
- Spatial information is retained through the formation of a
spatial grid and/or a spatial proximity network, which
is used to:
- identify spatial genes
- extract continuous spatial-expression patterns
- identify discrete spatial domains using HMRF
- explore cell-type/cell-type spatial interaction enrichment or depletion
- calculate spatially increased ligand-receptor expression in cells of interacting cell type pairs
- find interaction changed genes (ICG): genes that change expression in one cell type due to interaction with a neighboring cell type
- Giotto provides a number of options to visualize both 2D and 3D data and the outcome of Giotto can be interactively explored using Giotto Viewer, which allows you to overlay the obtained results with raw or additional images of the profiled tissue section(s).
Make sure to check out the Datasets section to see examples of the Giotto workflow.
HOWTOs
Giotto provides a lot of analyses, visualizations and other options to facilitate your spatial dataset analysis. We are working on providing easy-to-understand examples or tutorials, but if anything is not clear or if there is something you would like to see in particular, then do not hesitate to contact us.
Giotto tips & tricks
- Different ways of subsetting Giotto results?
- How to create global instructions and show or save your created plots?
- Different ways to visualize your spatial data?
- How to test and store multiple parameters or analyses?
- Visualize spatial data with voronoi plots
-
Adding and working with images in
Giotto
- Working with the Giotto class
Giotto analyses [work in progress]
- 0. Install a Giotto environment (optional)
- 1. Create a Giotto object
- 2. Process and filter a Giotto object
- 3. Dimension reduction
- 4. Cluster cells or spots
- 5. Identify differentially expressed genes
- 6. Annotate clusters
- 7. Cell-type enrichment or deconvolution per spot
-
8. Create a Spatial grid
or Network
- 9. Find genes with a spatially coherent gene expression pattern
- 10. Identify genes that are spatially co-expressed
- 11. Explore spatial domains with HMRF
- 12. Calculate spatial cell-cell interaction enrichment
- 13. Find cell-cell interaction changed genes (ICG)
- 14. Identify enriched or depleted ligand-receptor interactions in hetero and homo-typic cell interactions
- Export Giotto results to use in Giotto viewer