#> Warning: This tutorial was written with Giotto version 1.0.3, your version is
#> 1.1.2.This is a more recent version and results should be reproducible
library(Giotto)
# 1. set working directory
#results_folder = '/path/to/directory/'
results_folder = '/Volumes/Ruben_Seagate/Dropbox (Personal)/Projects/GC_lab/Ruben_Dries/190225_spatial_package/Results/Visium/Brain/201226_results//'
# 2. set giotto python path
# set python path to your preferred python version path
# set python path to NULL if you want to automatically install (only the 1st time) and use the giotto miniconda environment
python_path = NULL
if(is.null(python_path)) {
installGiottoEnvironment()
}Dataset explanation
10X genomics recently launched a new platform to obtain spatial expression data using a Visium Spatial Gene Expression slide.
The Visium brain data to run this tutorial can be found here
Visium technology: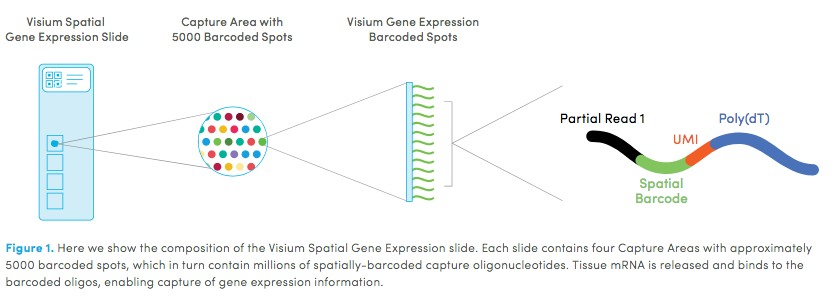
High resolution png from original tissue:
Part 1: Giotto global instructions and preparations
## create instructions
instrs = createGiottoInstructions(save_dir = results_folder,
save_plot = TRUE,
show_plot = FALSE)
## provide path to visium folder
#data_path = '/path/to/Brain_data/'
data_path = '/Volumes/Ruben_Seagate/Dropbox (Personal)/Projects/GC_lab/Ruben_Dries/190225_spatial_package/Data/Visium_data/Brain_data/'part 2: Create Giotto object & process data
## directly from visium folder
visium_brain = createGiottoVisiumObject(visium_dir = data_path, expr_data = 'raw',
png_name = 'tissue_lowres_image.png',
gene_column_index = 2, instructions = instrs)
## update and align background image
# problem: image is not perfectly aligned
spatPlot(gobject = visium_brain, cell_color = 'in_tissue', show_image = T, point_alpha = 0.7,
save_param = list(save_name = '2_a_spatplot_image'))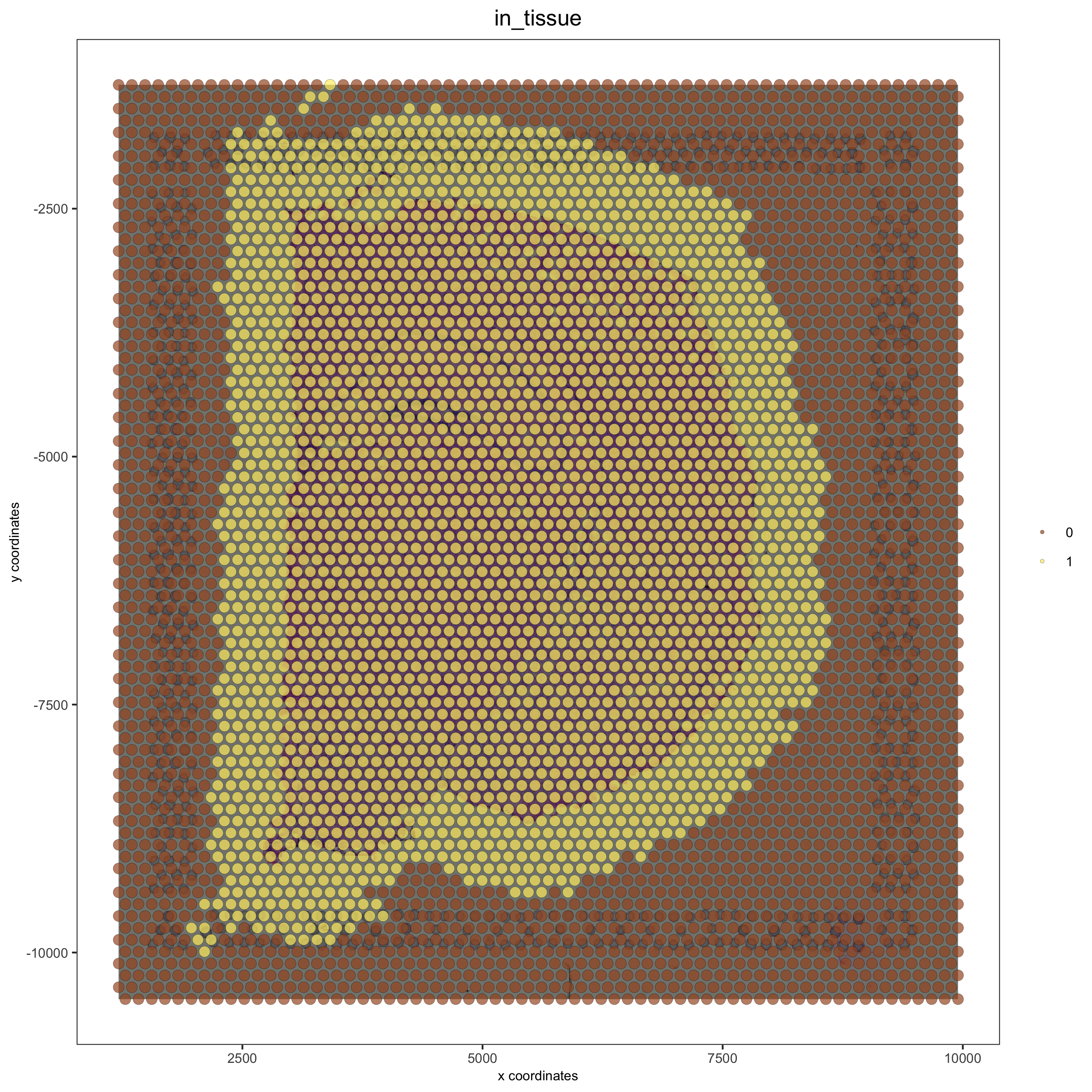
# check name
showGiottoImageNames(visium_brain) # "image" is the default name
# adjust parameters to align image (iterative approach)
visium_brain = updateGiottoImage(visium_brain, image_name = 'image',
xmax_adj = 1300, xmin_adj = 1200,
ymax_adj = 1100, ymin_adj = 1000)
# now it's aligned
spatPlot(gobject = visium_brain, cell_color = 'in_tissue', show_image = T, point_alpha = 0.7,
save_param = list(save_name = '2_b_spatplot_image_adjusted'))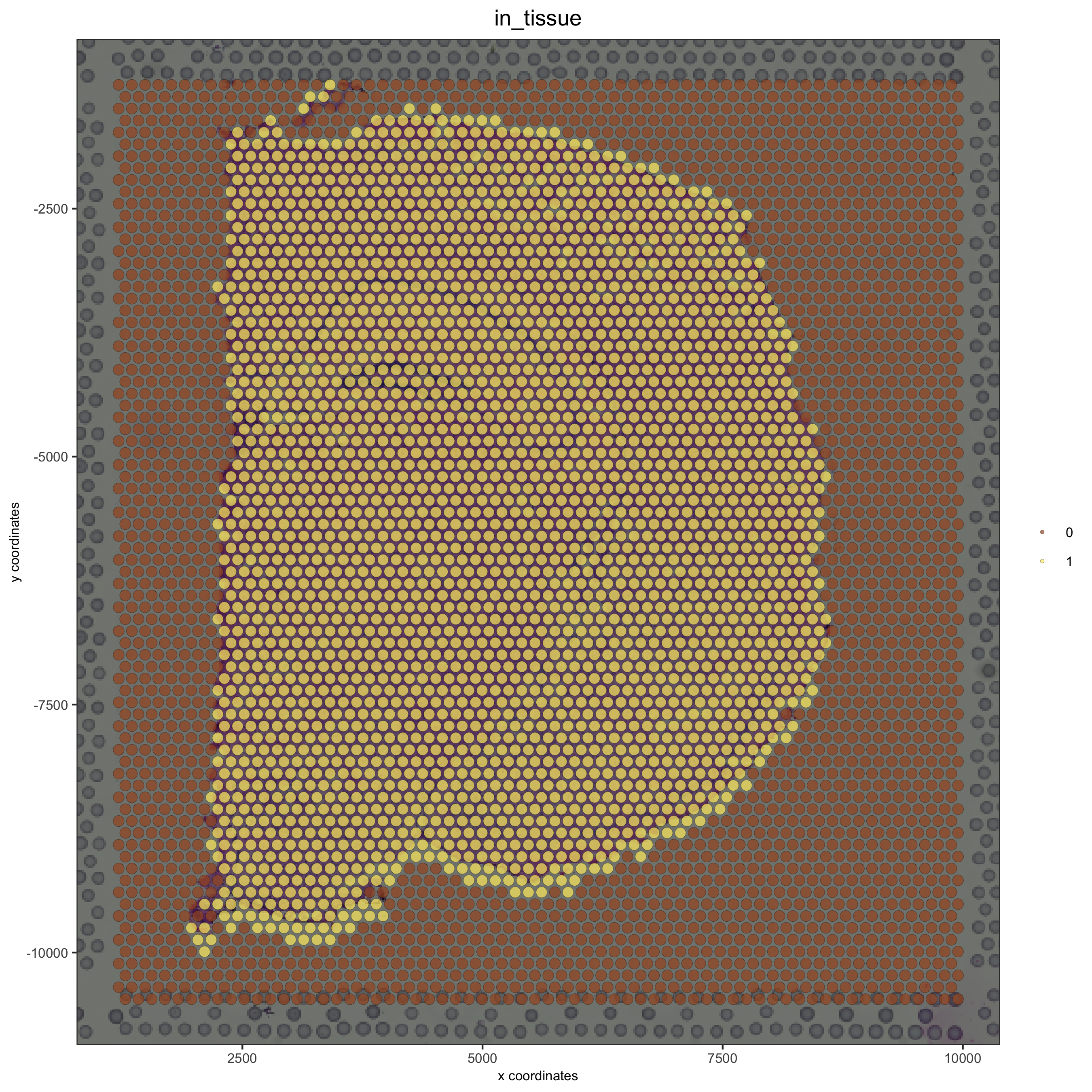
## check metadata
pDataDT(visium_brain)
## compare in tissue with provided jpg
spatPlot(gobject = visium_brain, cell_color = 'in_tissue', point_size = 2,
cell_color_code = c('0' = 'lightgrey', '1' = 'blue'),
save_param = list(save_name = '2_c_in_tissue'))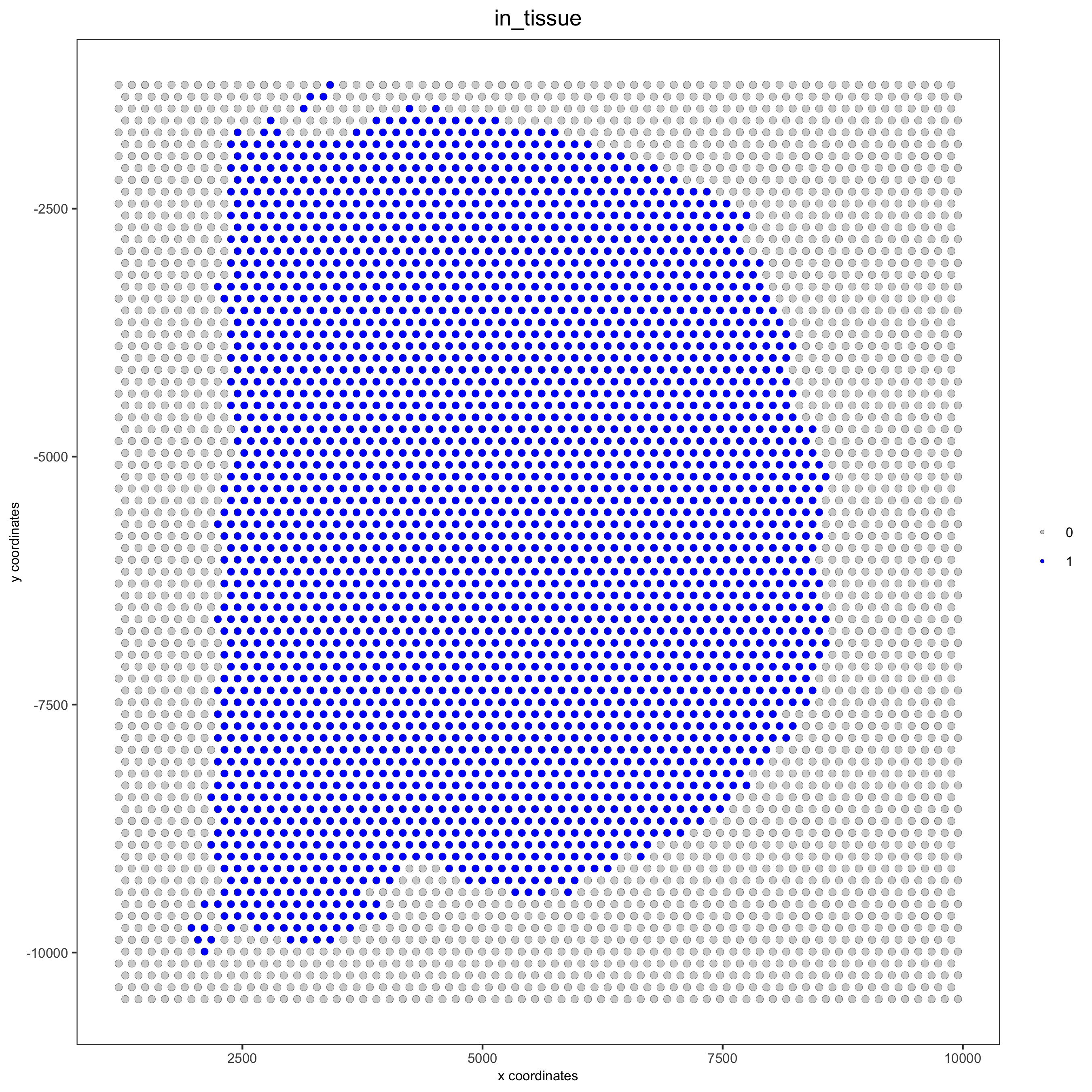
## subset on spots that were covered by tissue
metadata = pDataDT(visium_brain)
in_tissue_barcodes = metadata[in_tissue == 1]$cell_ID
visium_brain = subsetGiotto(visium_brain, cell_ids = in_tissue_barcodes)
## filter
visium_brain <- filterGiotto(gobject = visium_brain,
expression_threshold = 1,
gene_det_in_min_cells = 50,
min_det_genes_per_cell = 1000,
expression_values = c('raw'),
verbose = T)
## normalize
visium_brain <- normalizeGiotto(gobject = visium_brain, scalefactor = 6000, verbose = T)
## add gene & cell statistics
visium_brain <- addStatistics(gobject = visium_brain)
## visualize
spatPlot2D(gobject = visium_brain, show_image = T, point_alpha = 0.7,
save_param = list(save_name = '2_d_spatial_locations'))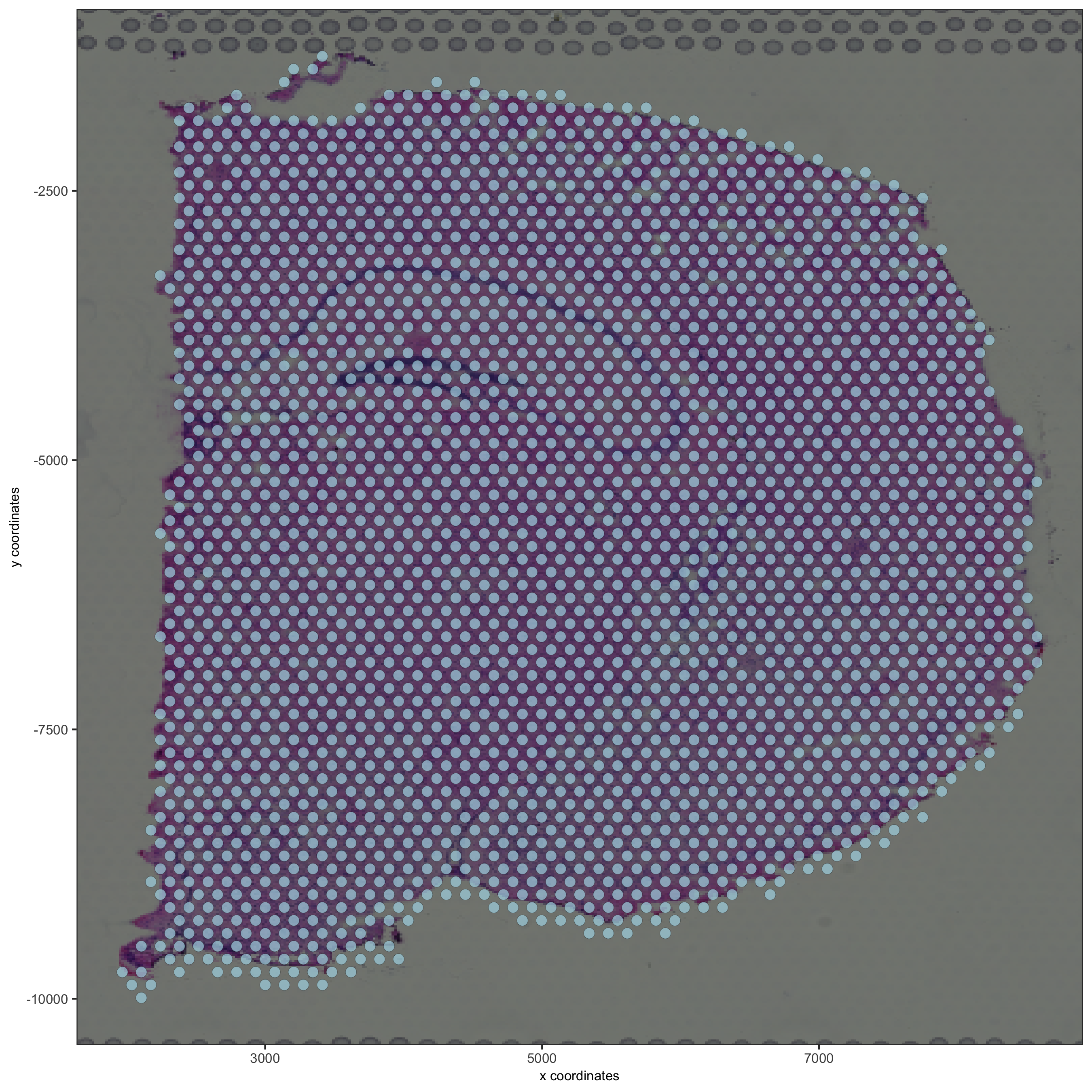
spatPlot2D(gobject = visium_brain, show_image = T, point_alpha = 0.7,
cell_color = 'nr_genes', color_as_factor = F,
save_param = list(save_name = '2_e_nr_genes'))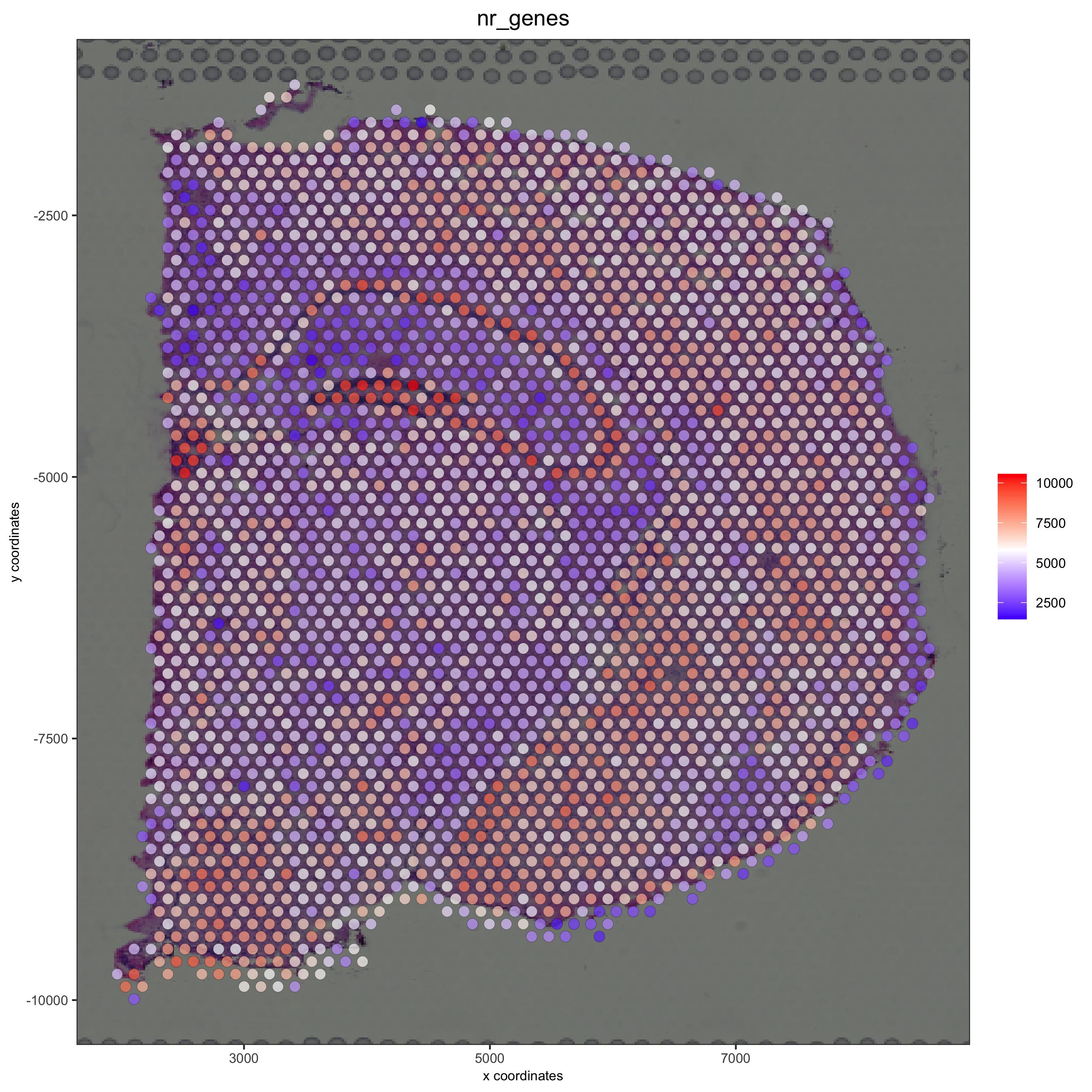
part 3: dimension reduction
## highly variable genes (HVG)
visium_brain <- calculateHVG(gobject = visium_brain,
save_param = list(save_name = '3_a_HVGplot'))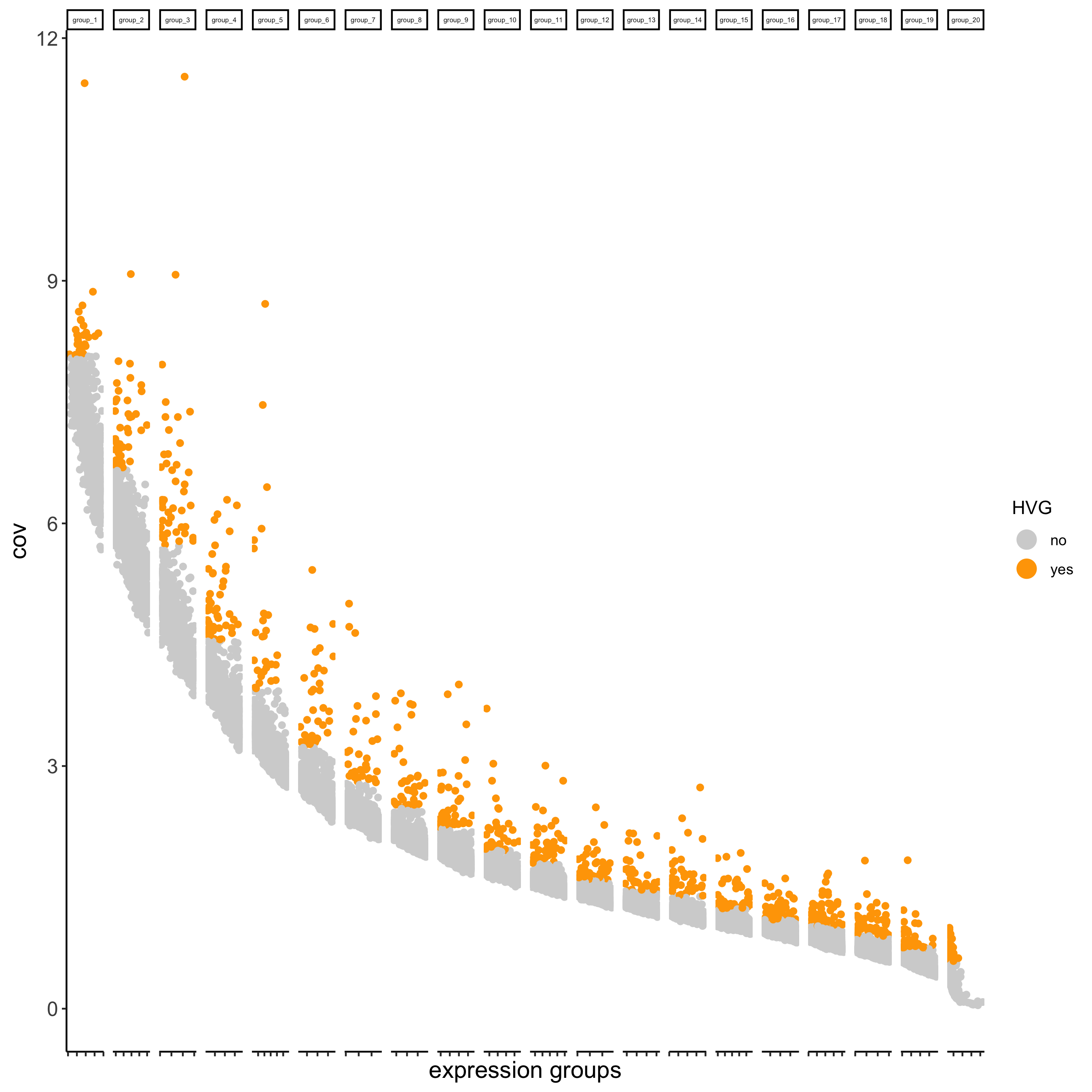
## run PCA on expression values (default)
gene_metadata = fDataDT(visium_brain)
featgenes = gene_metadata[hvg == 'yes' & perc_cells > 3 & mean_expr_det > 0.4]$gene_ID
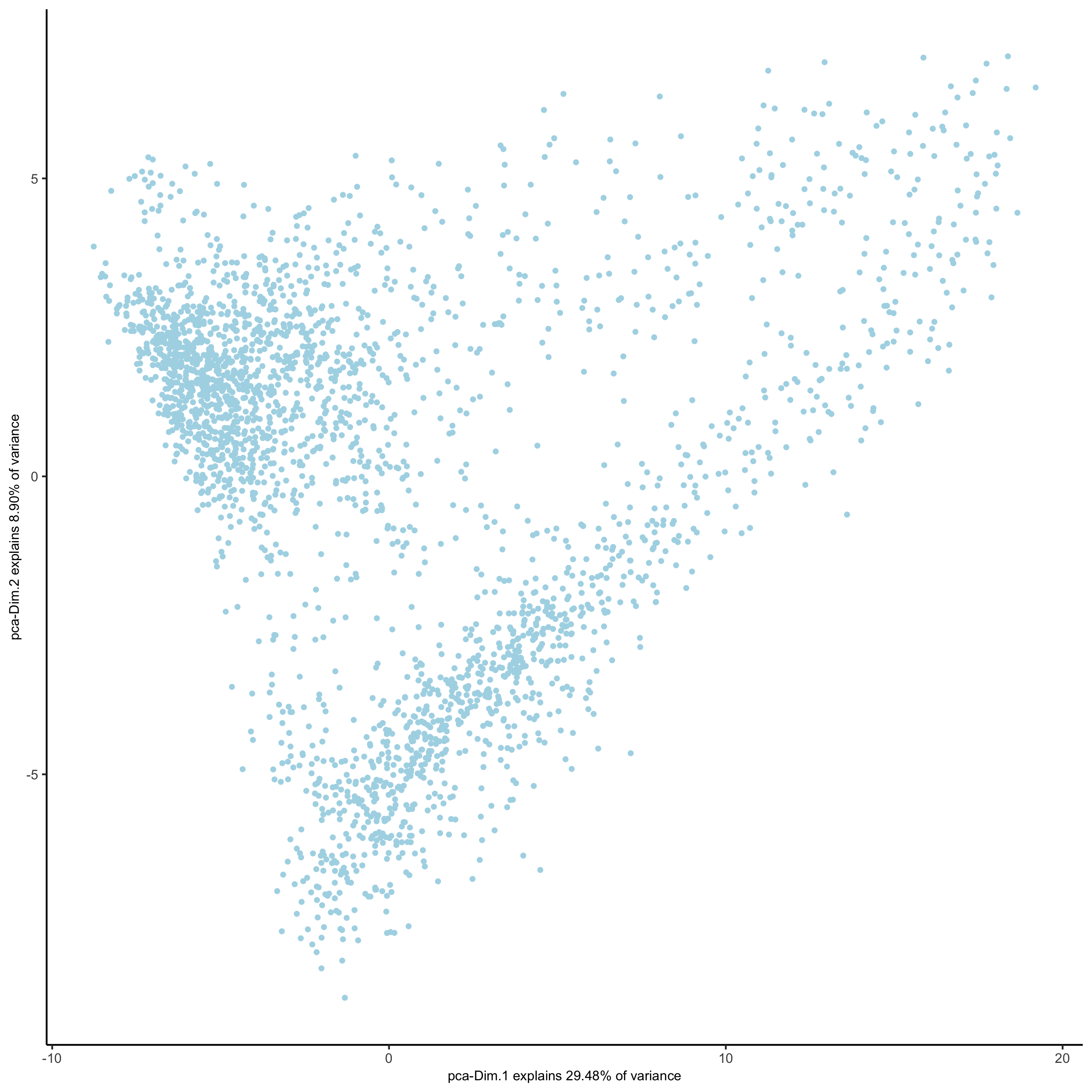
visium_brain <- runPCA(gobject = visium_brain,
genes_to_use = featgenes,
scale_unit = F, center = T,
method="factominer")
screePlot(visium_brain, ncp = 30, save_param = list(save_name = '3_b_screeplot'))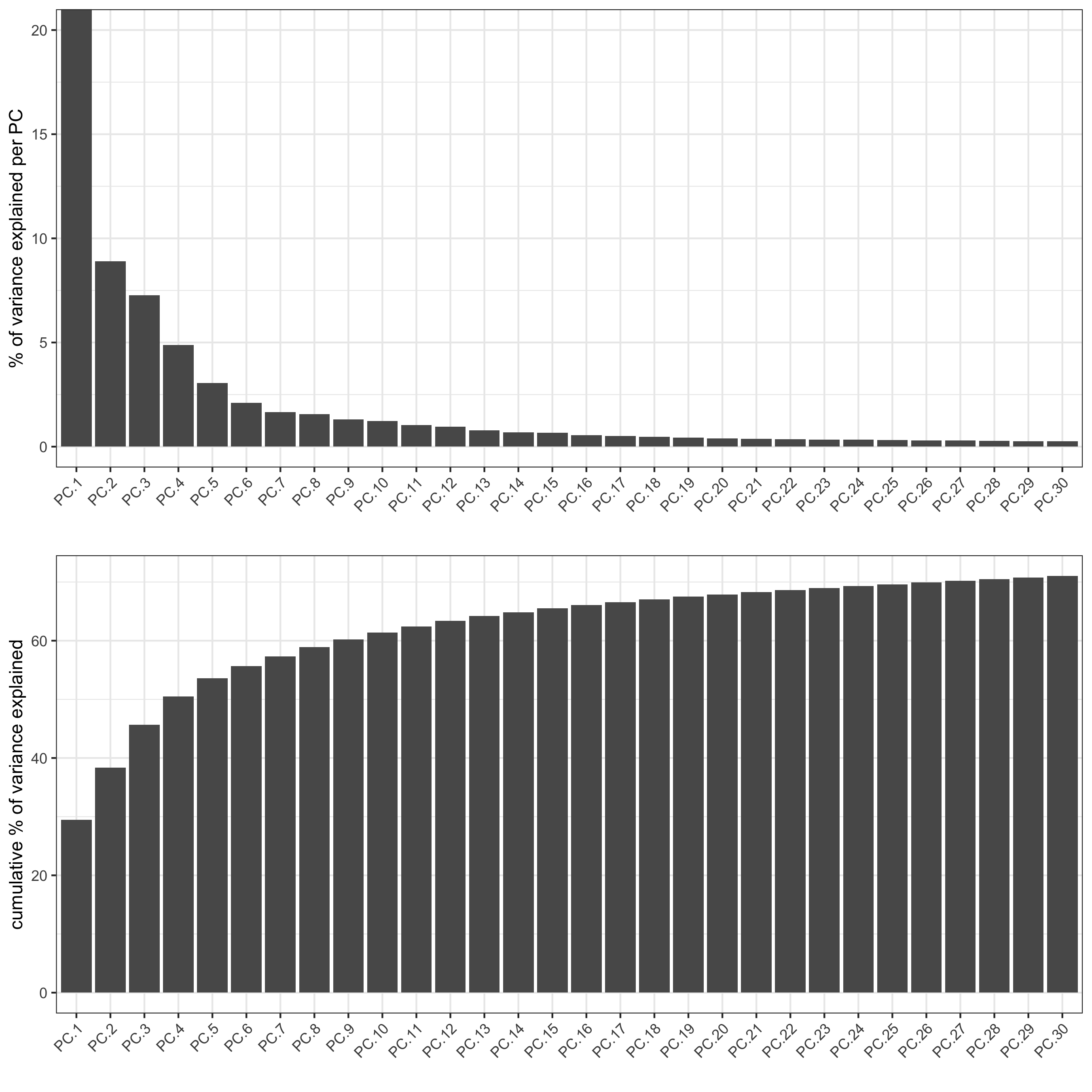
## run UMAP and tSNE on PCA space (default)
visium_brain <- runUMAP(visium_brain, dimensions_to_use = 1:10)
plotUMAP(gobject = visium_brain,
save_param = list(save_name = '3_d_UMAP_reduction'))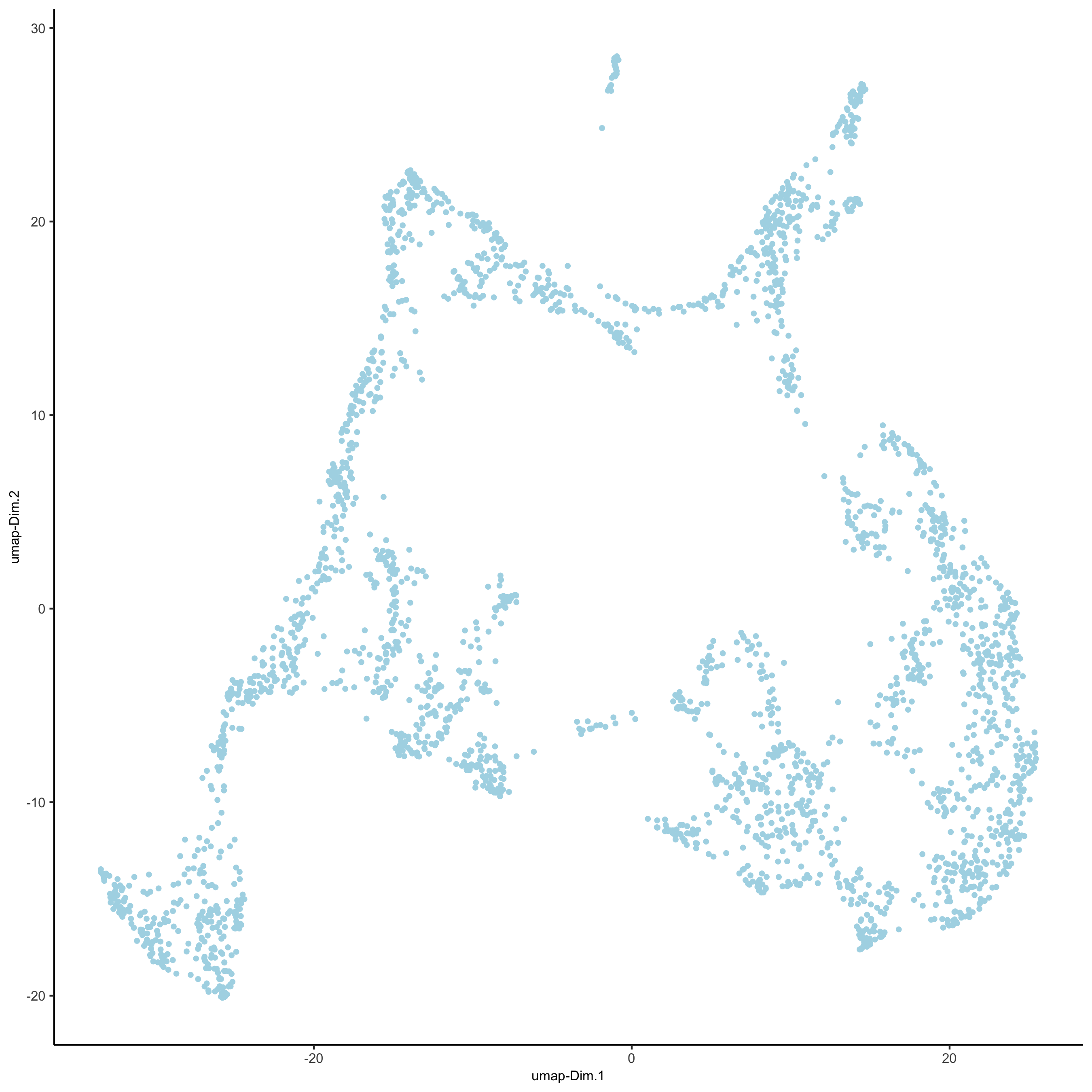
visium_brain <- runtSNE(visium_brain, dimensions_to_use = 1:10)
plotTSNE(gobject = visium_brain,
save_param = list(save_name = '3_e_tSNE_reduction'))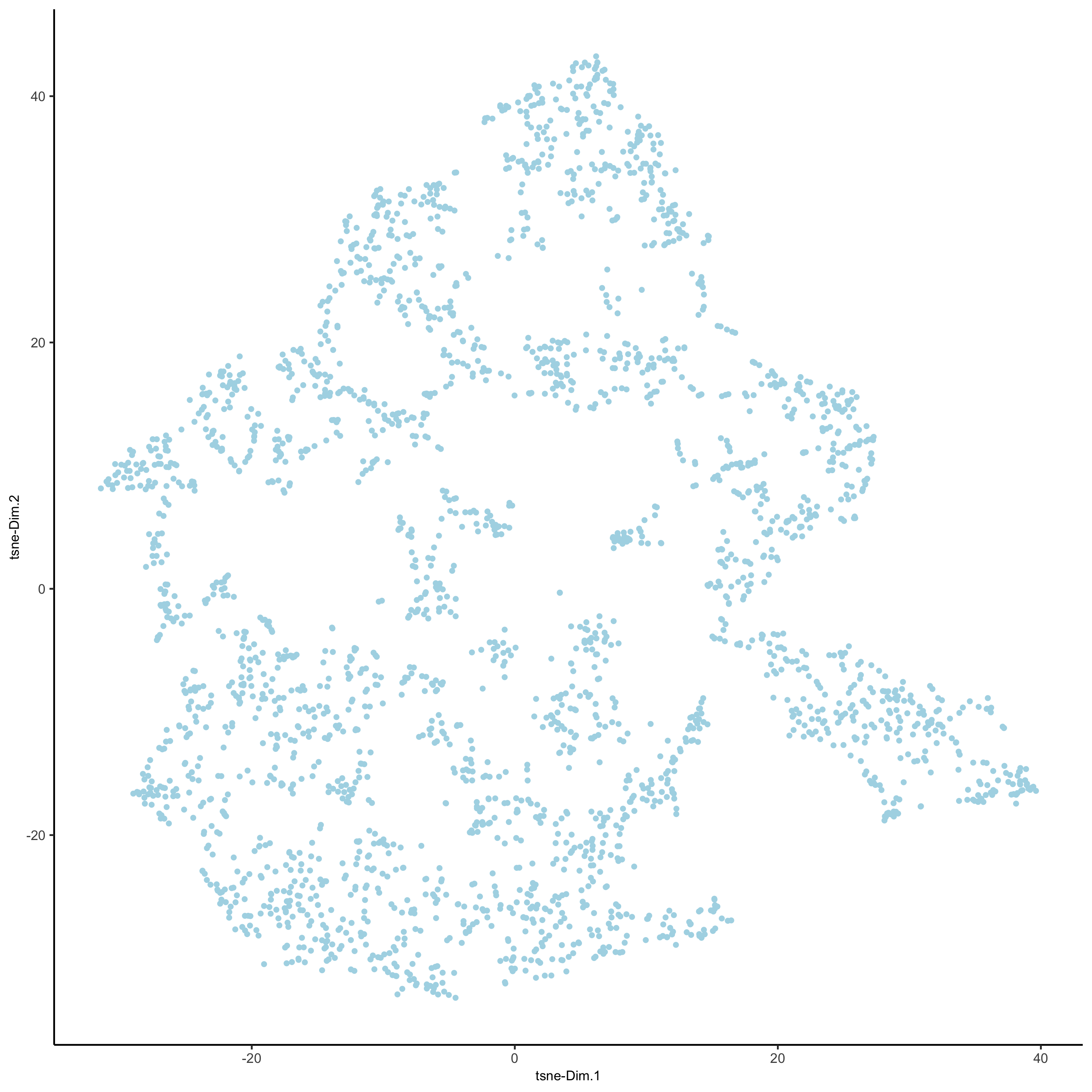
part 4: cluster
## sNN network (default)
visium_brain <- createNearestNetwork(gobject = visium_brain, dimensions_to_use = 1:10, k = 15)
## Leiden clustering
visium_brain <- doLeidenCluster(gobject = visium_brain, resolution = 0.4, n_iterations = 1000)
plotUMAP(gobject = visium_brain,
cell_color = 'leiden_clus', show_NN_network = T, point_size = 2.5,
save_param = list(save_name = '4_a_UMAP_leiden'))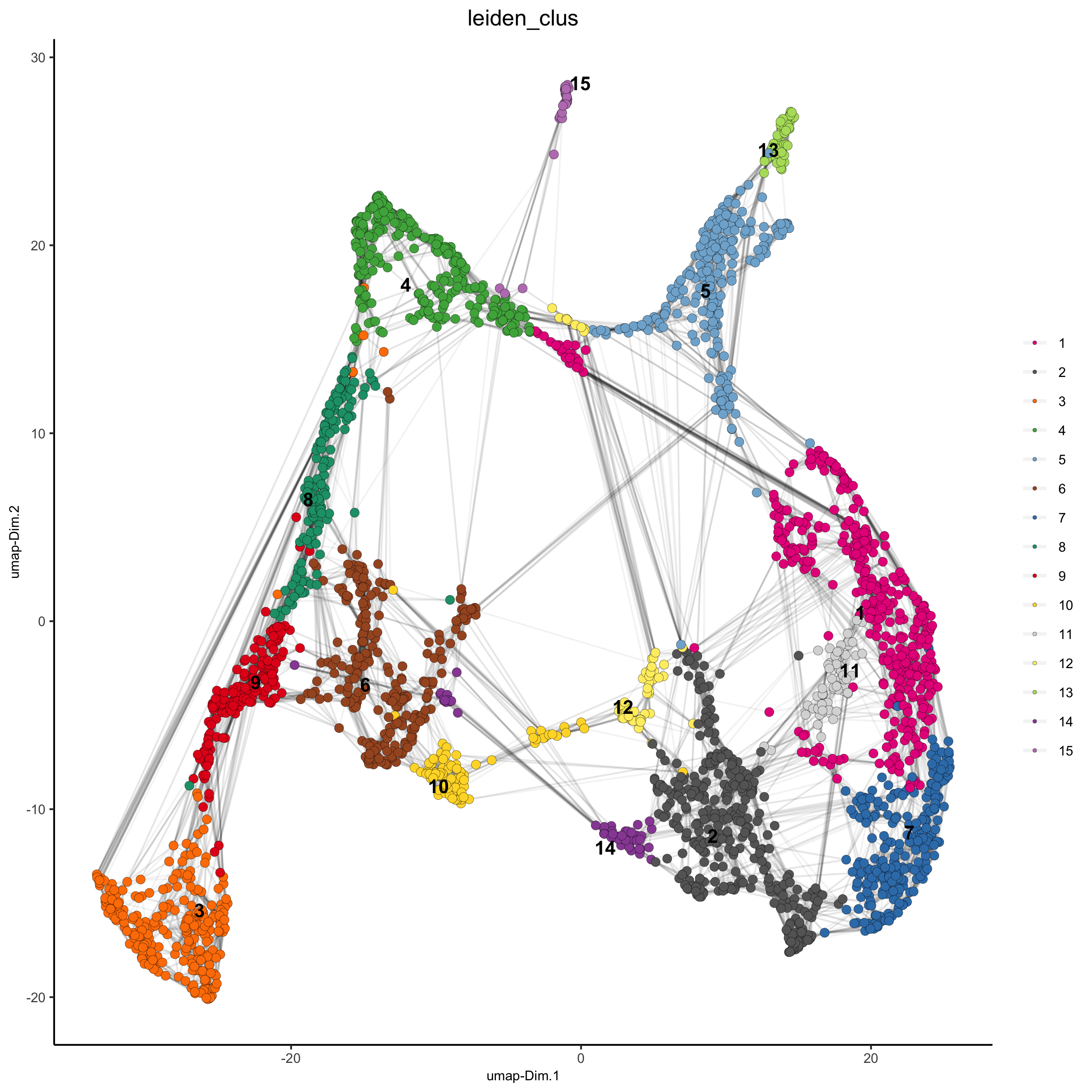
part 5: co-visualize
5.1 whole dataset
# expression and spatial
spatDimPlot(gobject = visium_brain, cell_color = 'leiden_clus',
dim_point_size = 2, spat_point_size = 2.5,
save_param = list(save_name = '5_a_covis_leiden'))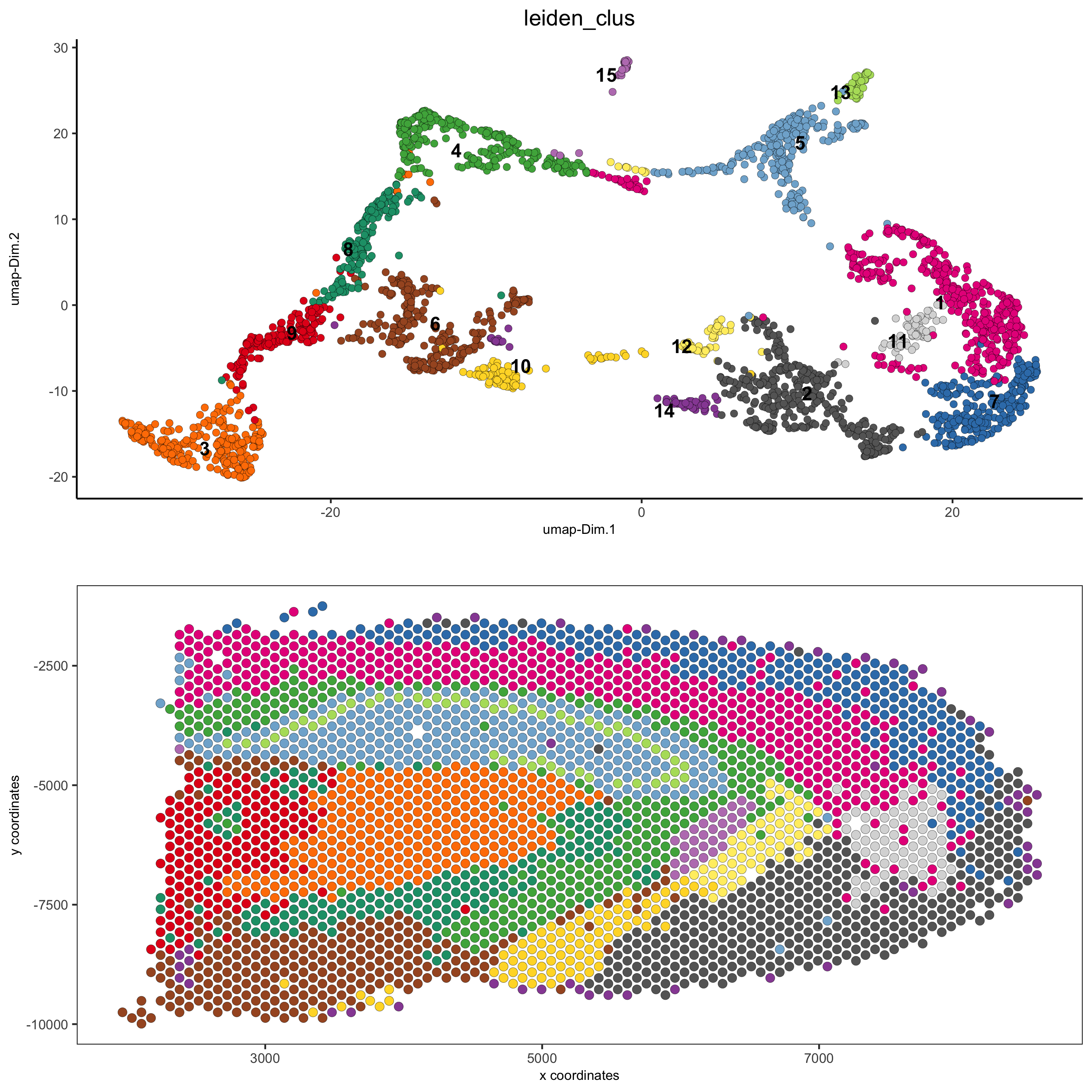
spatDimPlot(gobject = visium_brain, cell_color = 'nr_genes', color_as_factor = F,
dim_point_size = 2, spat_point_size = 2.5,
save_param = list(save_name = '5_b_nr_genes'))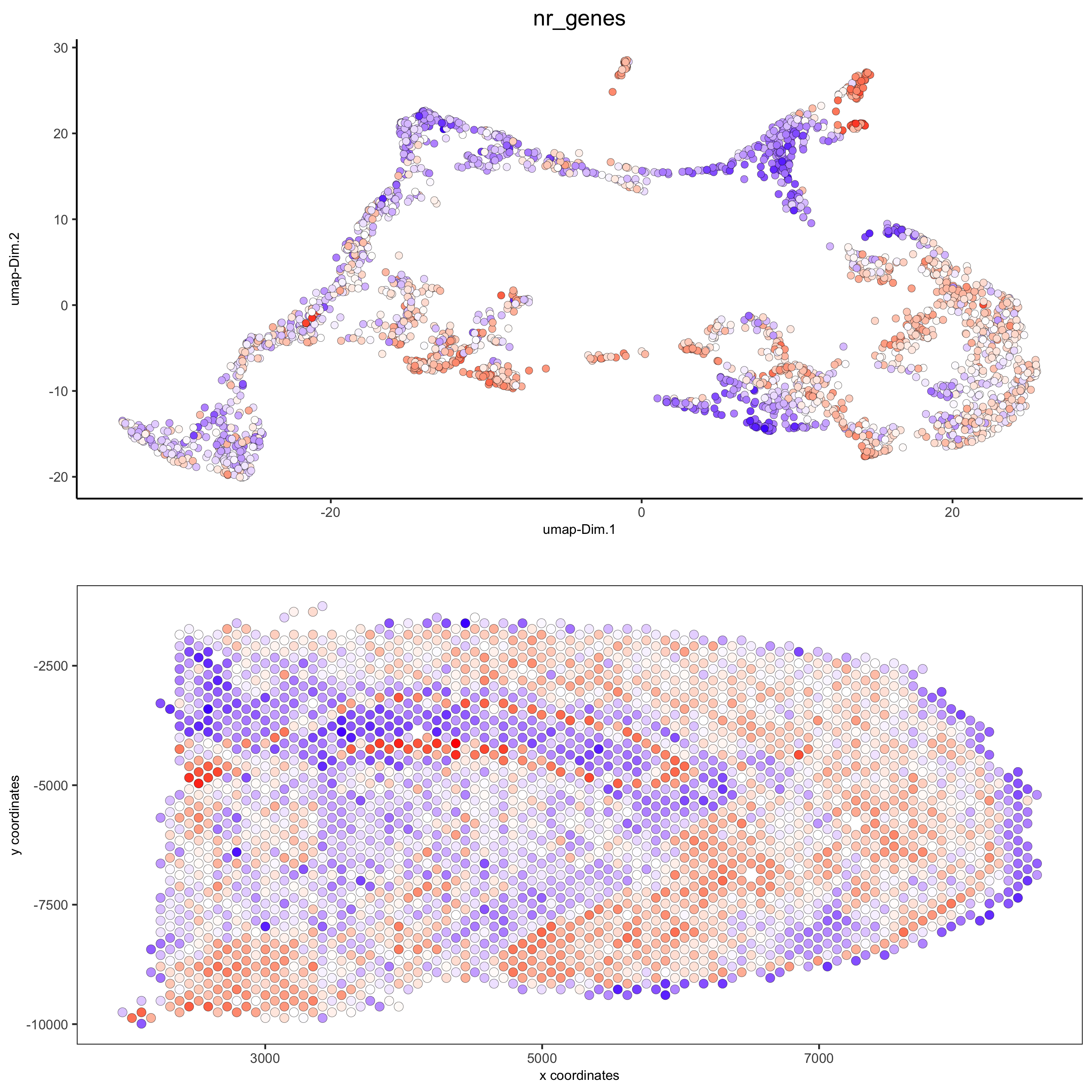
5.2 subset dataset on DG region
DG_subset = subsetGiottoLocs(visium_brain,
x_max = 6500, x_min = 3000,
y_max = -2500, y_min = -5500,
return_gobject = TRUE)
spatDimPlot(gobject = DG_subset,
cell_color = 'leiden_clus', spat_point_size = 5,
save_param = list(save_name = '5_c_DEG_subset'))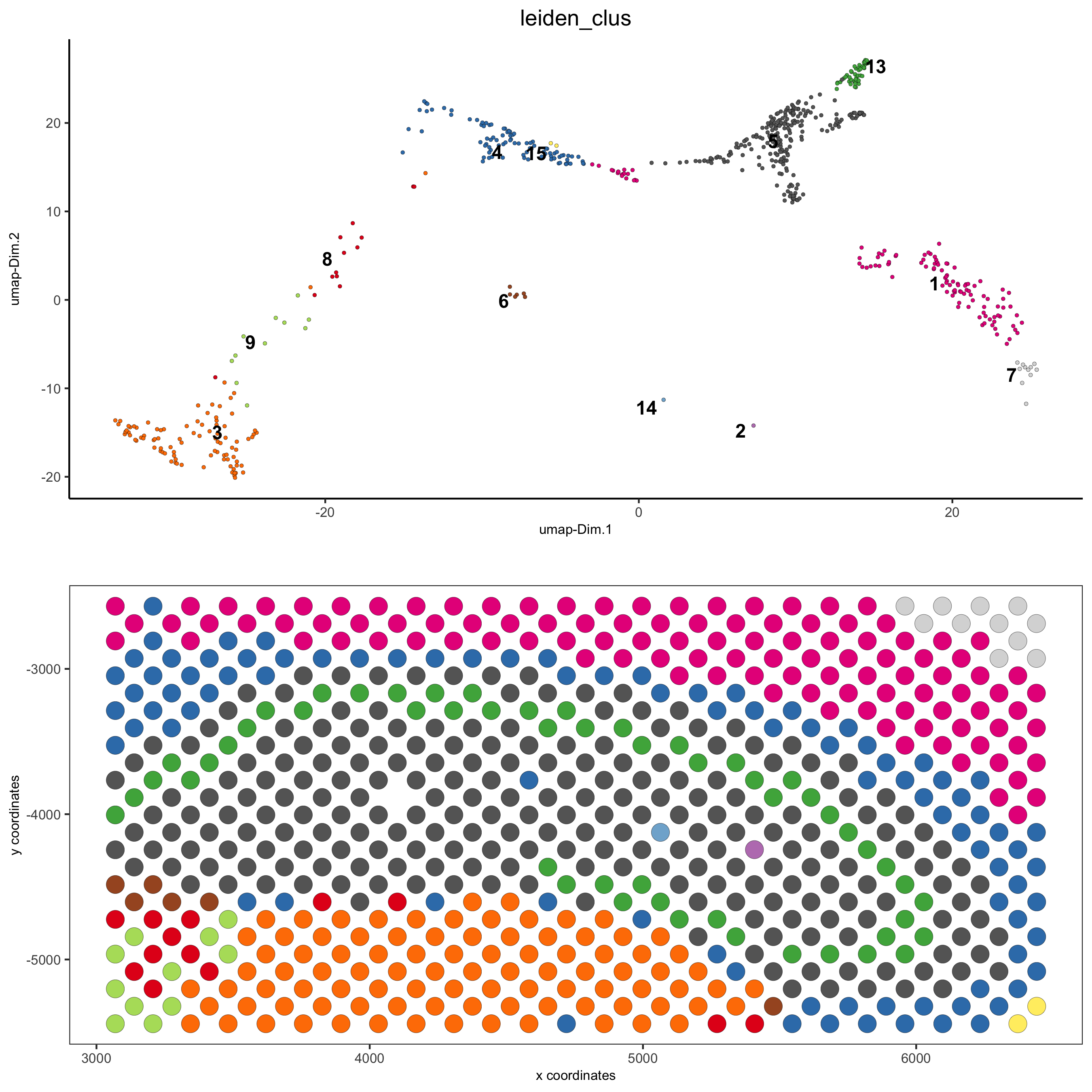
part 6: cell type marker gene detection
gini
gini_markers_subclusters = findMarkers_one_vs_all(gobject = visium_brain,
method = 'gini',
expression_values = 'normalized',
cluster_column = 'leiden_clus',
min_genes = 20,
min_expr_gini_score = 0.5,
min_det_gini_score = 0.5)
topgenes_gini = gini_markers_subclusters[, head(.SD, 2), by = 'cluster']$genes
# violinplot
violinPlot(visium_brain, genes = unique(topgenes_gini), cluster_column = 'leiden_clus',
strip_text = 8, strip_position = 'right',
save_param = list(save_name = '6_a_violinplot_gini', base_width = 5, base_height = 10))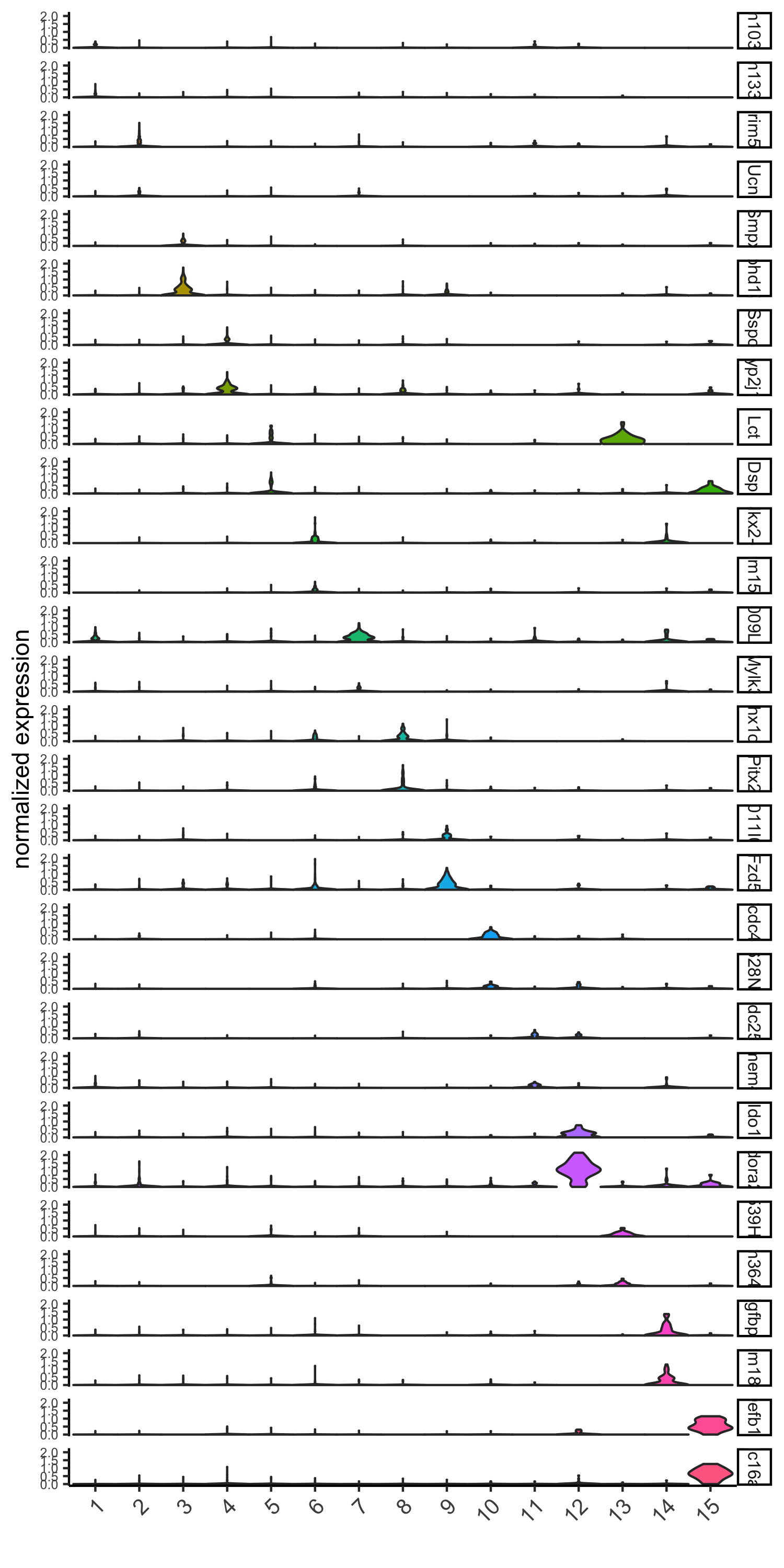
# cluster heatmap
plotMetaDataHeatmap(visium_brain, selected_genes = topgenes_gini,
metadata_cols = c('leiden_clus'),
x_text_size = 10, y_text_size = 10,
save_param = list(save_name = '6_b_metaheatmap_gini'))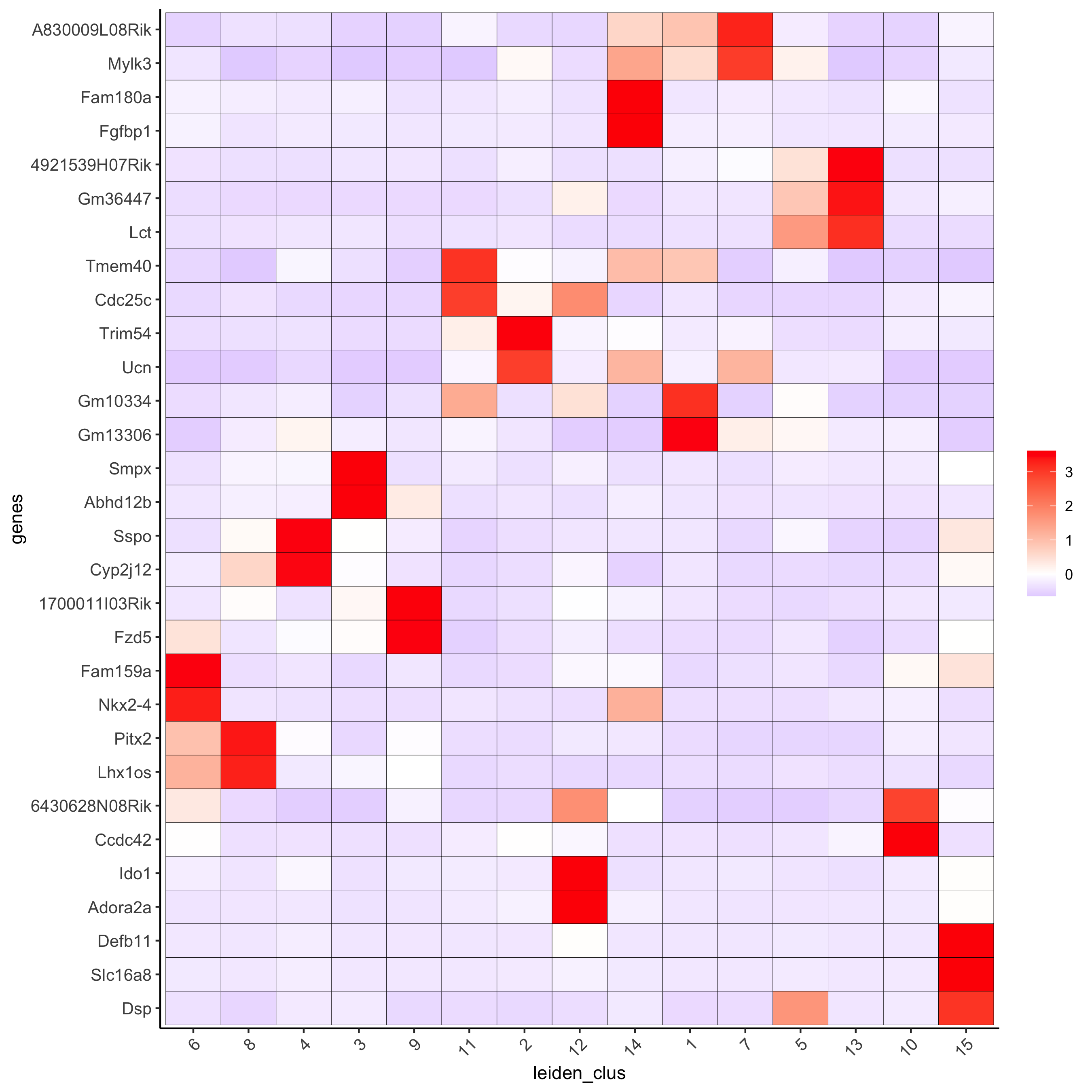
# umap plots
dimGenePlot2D(visium_brain, expression_values = 'scaled',
genes = gini_markers_subclusters[, head(.SD, 1), by = 'cluster']$genes,
cow_n_col = 3, point_size = 1,
save_param = list(save_name = '6_c_gini_umap', base_width = 8, base_height = 5))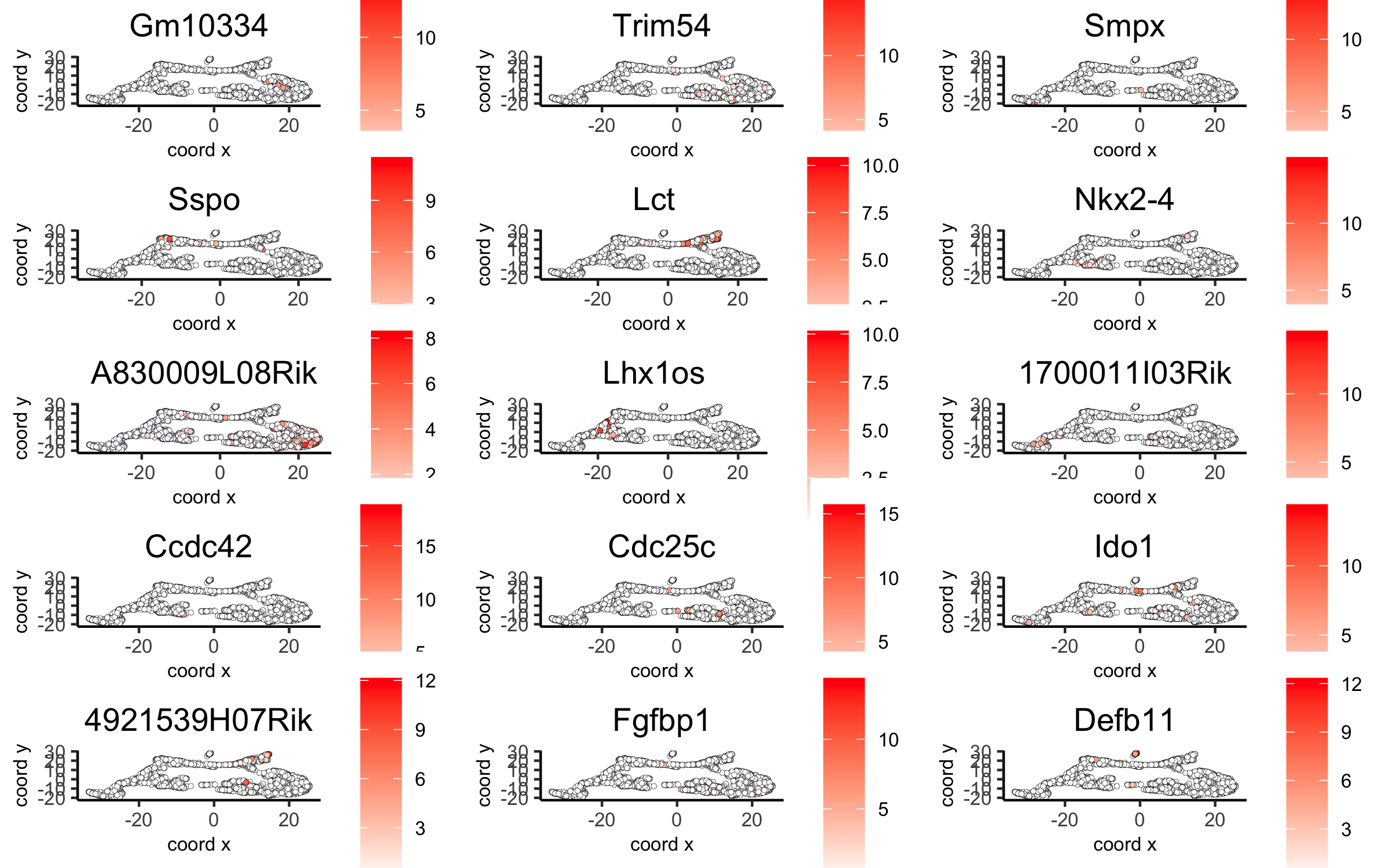
scran
scran_markers_subclusters = findMarkers_one_vs_all(gobject = visium_brain,
method = 'scran',
expression_values = 'normalized',
cluster_column = 'leiden_clus')
topgenes_scran = scran_markers_subclusters[, head(.SD, 2), by = 'cluster']$genes
# violinplot
violinPlot(visium_brain, genes = unique(topgenes_scran), cluster_column = 'leiden_clus',
strip_text = 10, strip_position = 'right',
save_param = list(save_name = '6_d_violinplot_scran', base_width = 5))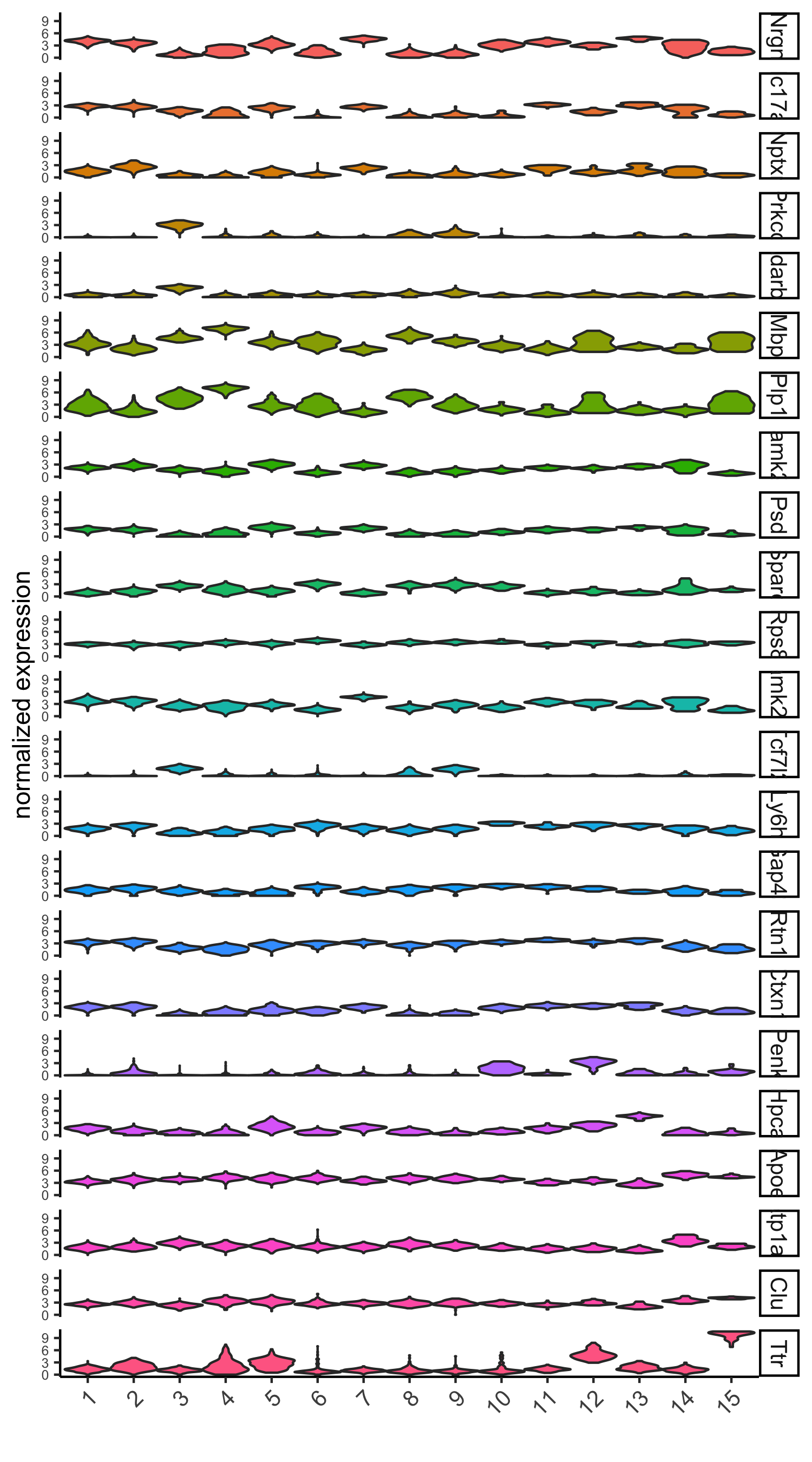
# cluster heatmap
plotMetaDataHeatmap(visium_brain, selected_genes = topgenes_scran,
metadata_cols = c('leiden_clus'),
save_param = list(save_name = '6_e_metaheatmap_scran'))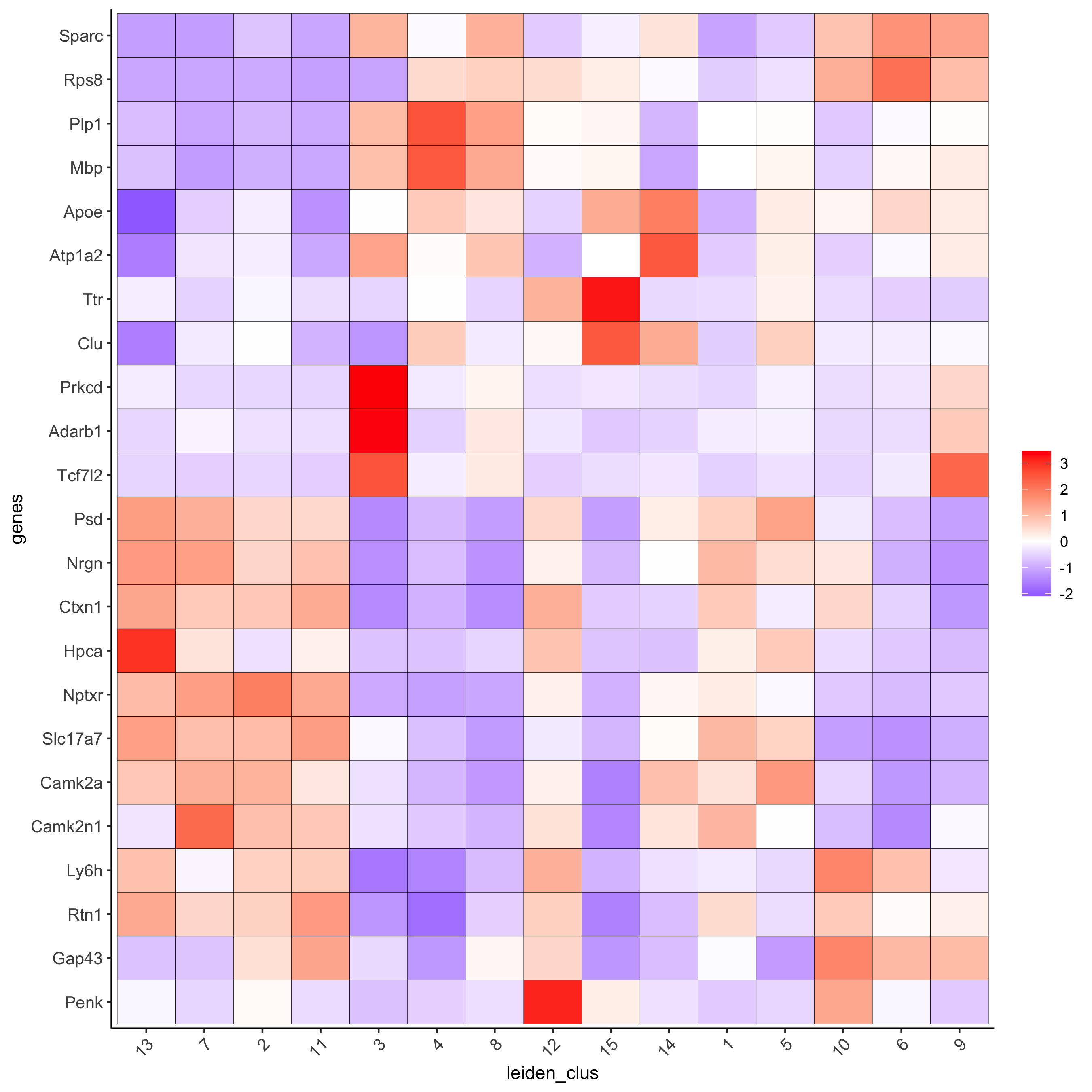
# umap plots
dimGenePlot(visium_brain, expression_values = 'scaled',
genes = scran_markers_subclusters[, head(.SD, 1), by = 'cluster']$genes,
cow_n_col = 3, point_size = 1,
save_param = list(save_name = '6_f_scran_umap', base_width = 8, base_height = 5))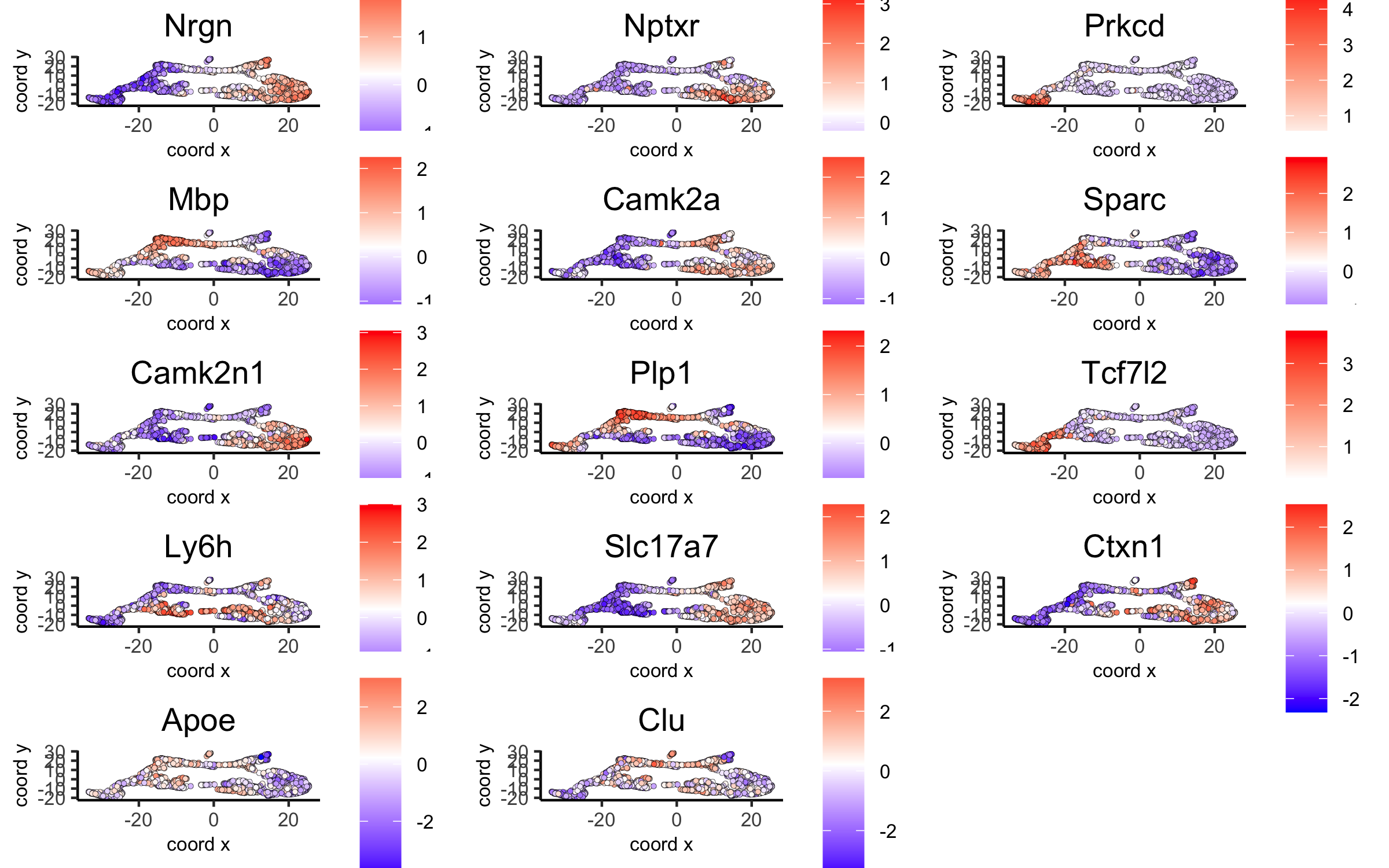
part 7: cell-type annotation
Visium spatial transcriptomics does not provide single-cell
resolution, making cell type annotation a harder problem. Giotto
provides 3 ways to calculate enrichment of specific cell-type signature
gene list:
- PAGE
- rank
- hypergeometric test
known markers for different mouse brain cell types:
Zeisel, A. et al. Molecular Architecture of the Mouse Nervous System.
Cell 174, 999-1014.e22 (2018). cell type signatures
combination of all marker genes identified in Zeisel et al
7.1 PAGE enrichment
# 1.1 create binary matrix of cell signature genes
# small example #
gran_markers = c("Nr3c2", "Gabra5", "Tubgcp2", "Ahcyl2",
"Islr2", "Rasl10a", "Tmem114", "Bhlhe22",
"Ntf3", "C1ql2")
oligo_markers = c("Efhd1", "H2-Ab1", "Enpp6", "Ninj2",
"Bmp4", "Tnr", "Hapln2", "Neu4",
"Wfdc18", "Ccp110")
di_mesench_markers = c("Cartpt", "Scn1a", "Lypd6b", "Drd5",
"Gpr88", "Plcxd2", "Cpne7", "Pou4f1",
"Ctxn2", "Wnt4")
signature_matrix = makeSignMatrixPAGE(sign_names = c('Granule_neurons',
'Oligo_dendrocytes',
'di_mesenchephalon'),
sign_list = list(gran_markers,
oligo_markers,
di_mesench_markers))
# 1.2 [shortcut] fully pre-prepared matrix for all cell types
sign_matrix_path = system.file("extdata", "sig_matrix.txt", package = 'Giotto')
brain_sc_markers = data.table::fread(sign_matrix_path)
sig_matrix = as.matrix(brain_sc_markers[,-1]); rownames(sig_matrix) = brain_sc_markers$Event
# 1.3 enrichment test with PAGE
# runSpatialEnrich() can also be used as a wrapper for all currently provided enrichment options
visium_brain = runPAGEEnrich(gobject = visium_brain, sign_matrix = sig_matrix)
# 1.4 heatmap of enrichment versus annotation (e.g. clustering result)
cell_types = colnames(sig_matrix)
plotMetaDataCellsHeatmap(gobject = visium_brain,
metadata_cols = 'leiden_clus',
value_cols = cell_types,
spat_enr_names = 'PAGE',
x_text_size = 8,
y_text_size = 8,
save_param = list(save_name="7_a_metaheatmap"))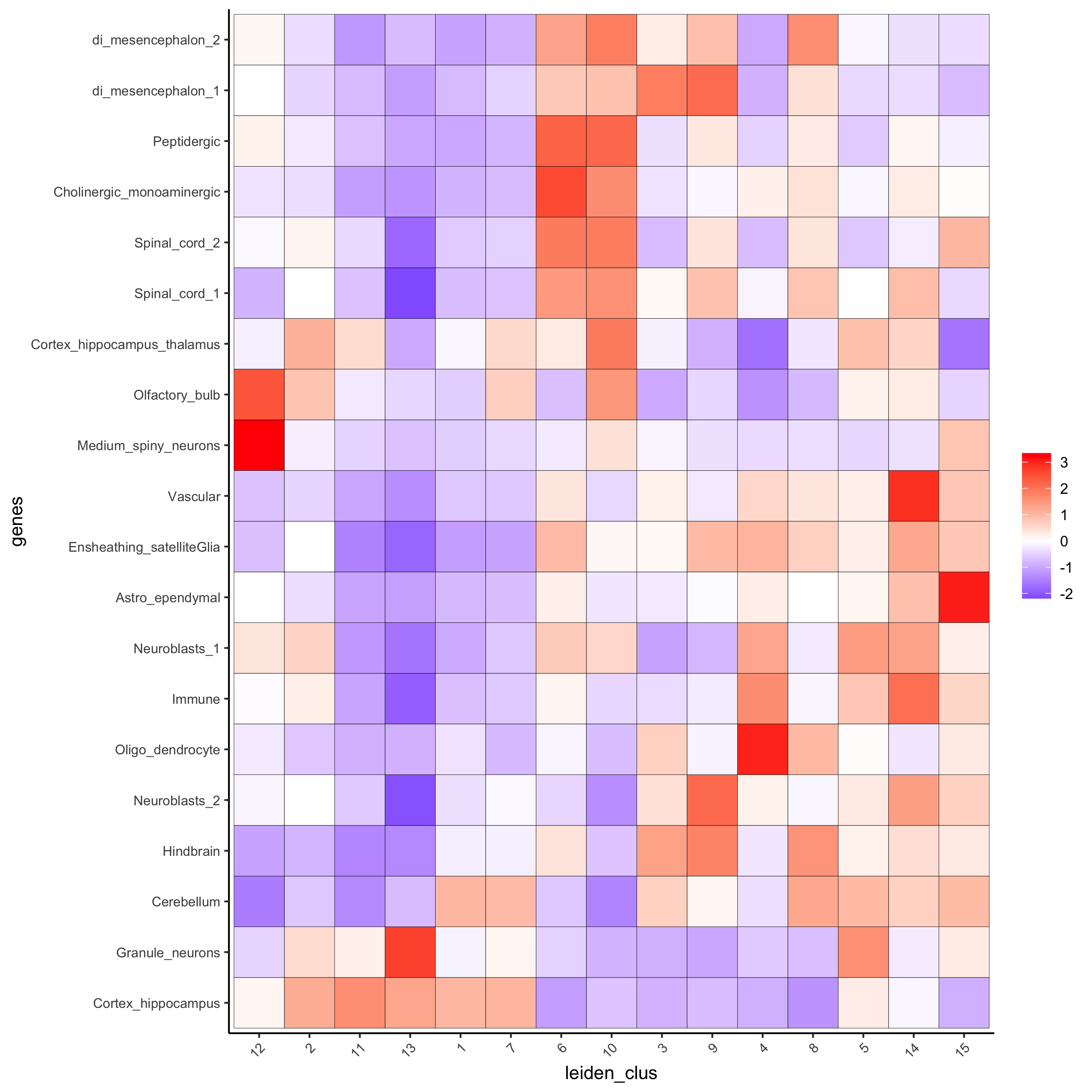
# 1.5 visualizations
cell_types_subset = colnames(sig_matrix)[1:10]
spatCellPlot(gobject = visium_brain,
spat_enr_names = 'PAGE',
cell_annotation_values = cell_types_subset,
cow_n_col = 4,coord_fix_ratio = NULL, point_size = 0.75,
save_param = list(save_name="7_b_spatcellplot_1"))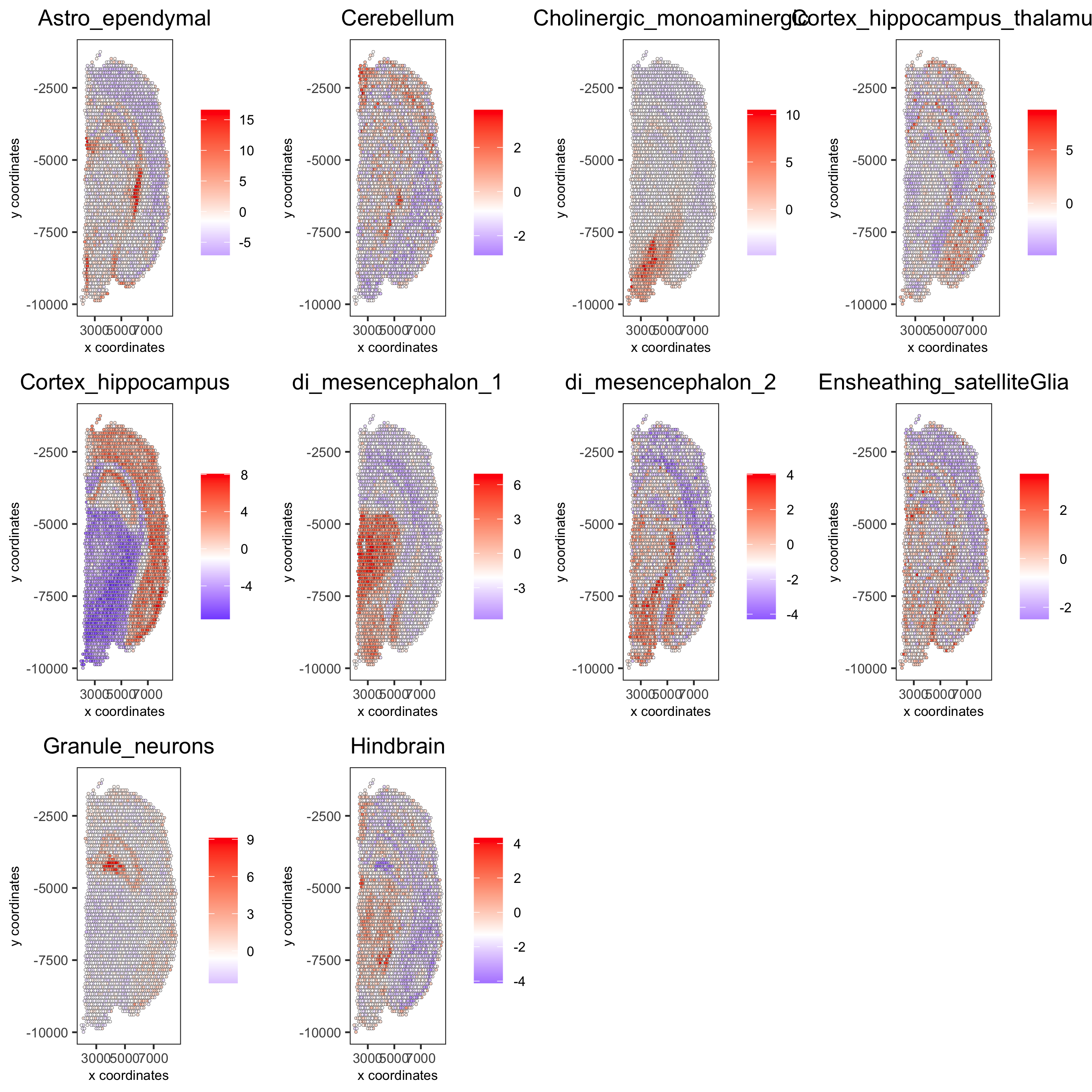
cell_types_subset = colnames(sig_matrix)[11:20]
spatCellPlot(gobject = visium_brain, spat_enr_names = 'PAGE',
cell_annotation_values = cell_types_subset, cow_n_col = 4,
coord_fix_ratio = NULL, point_size = 0.75,
save_param = list(save_name="7_c_spatcellplot_2"))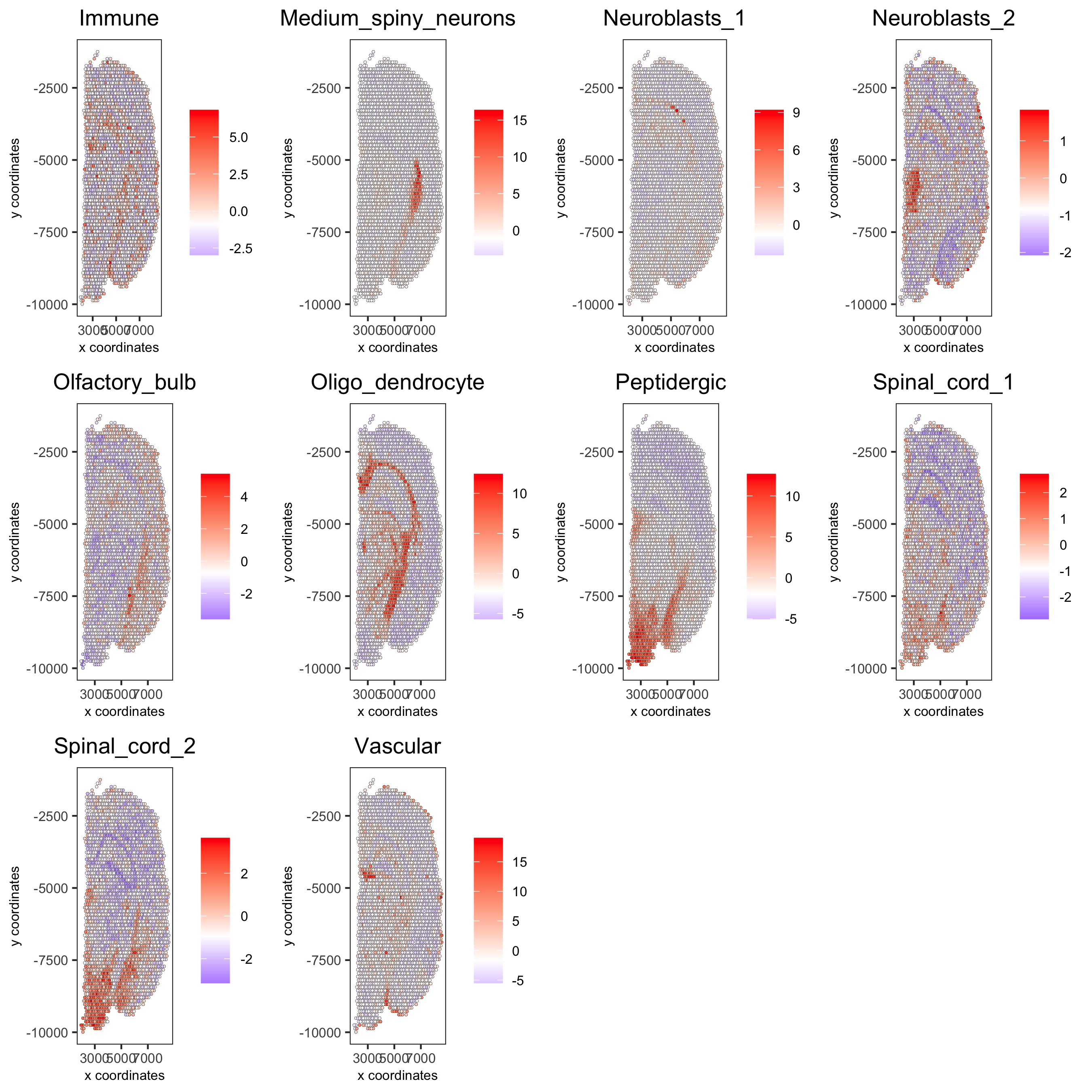
spatDimCellPlot(gobject = visium_brain,
spat_enr_names = 'PAGE',
cell_annotation_values = c('Cortex_hippocampus', 'Granule_neurons',
'di_mesencephalon_1', 'Oligo_dendrocyte','Vascular'),
cow_n_col = 1, spat_point_size = 1,
plot_alignment = 'horizontal',
save_param = list(save_name="7_d_spatDimCellPlot", base_width=7, base_height=10))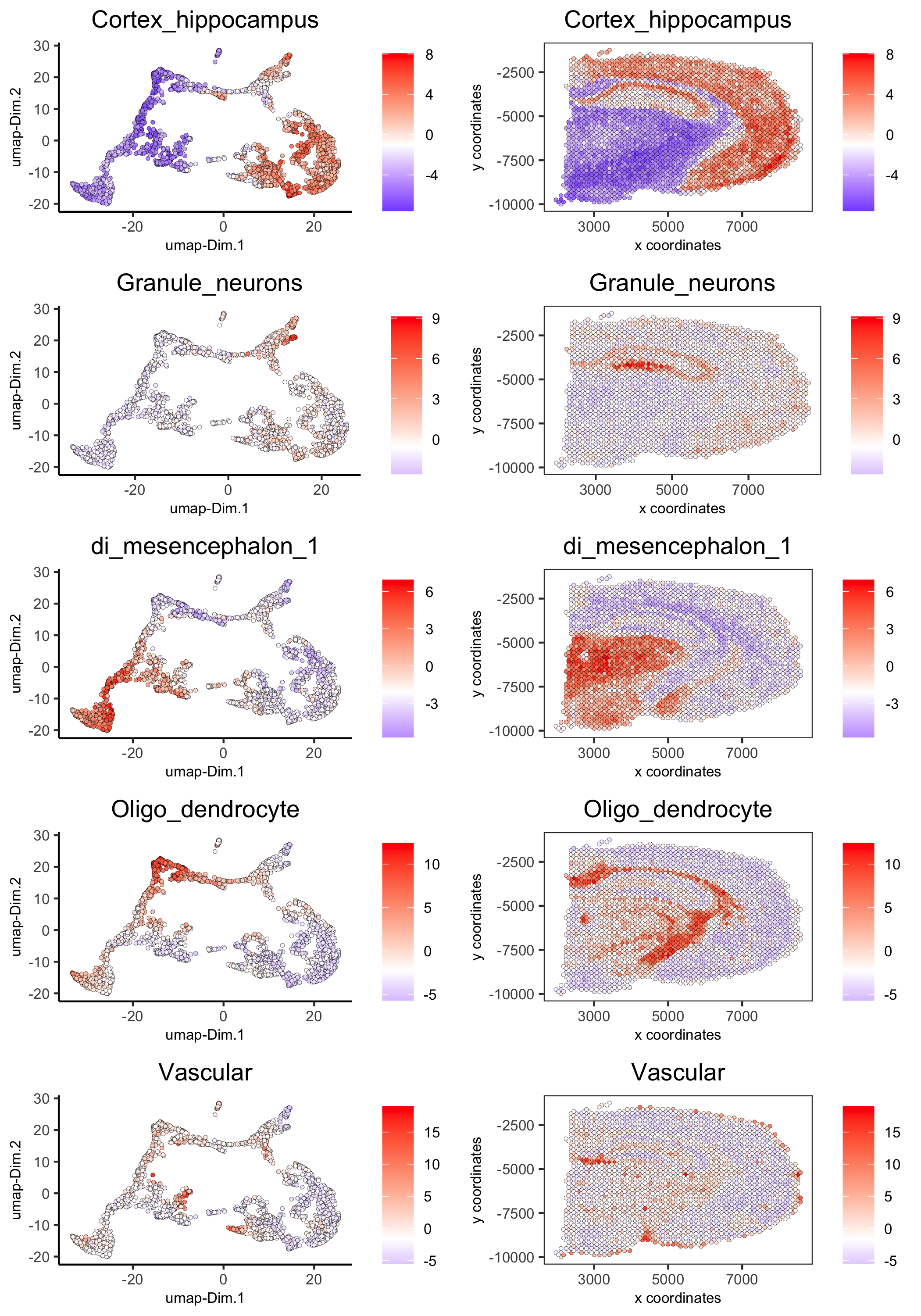
part 8: spatial grid
visium_brain <- createSpatialGrid(gobject = visium_brain,
sdimx_stepsize = 400,
sdimy_stepsize = 400,
minimum_padding = 0)
spatPlot(visium_brain, cell_color = 'leiden_clus', show_grid = T,
grid_color = 'red', spatial_grid_name = 'spatial_grid',
save_param = list(save_name = '8_grid'))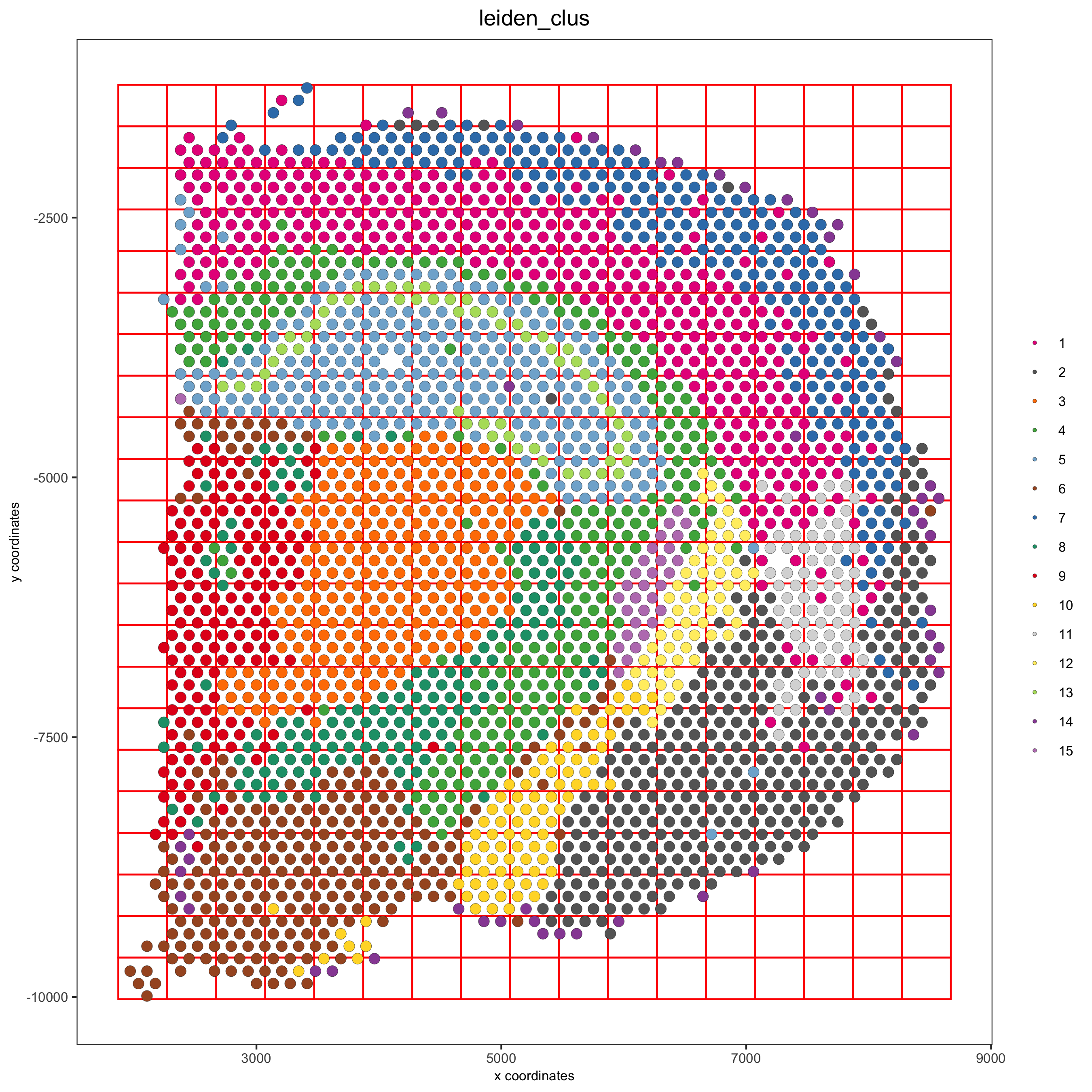
part 9: spatial network
visium_brain <- createSpatialNetwork(gobject = visium_brain,
method = 'kNN', k = 5,
maximum_distance_knn = 400,
name = 'spatial_network')
showNetworks(visium_brain)
spatPlot(gobject = visium_brain, show_network = T,
network_color = 'blue', spatial_network_name = 'spatial_network',
save_param = list(save_name = '9_a_knn_network'))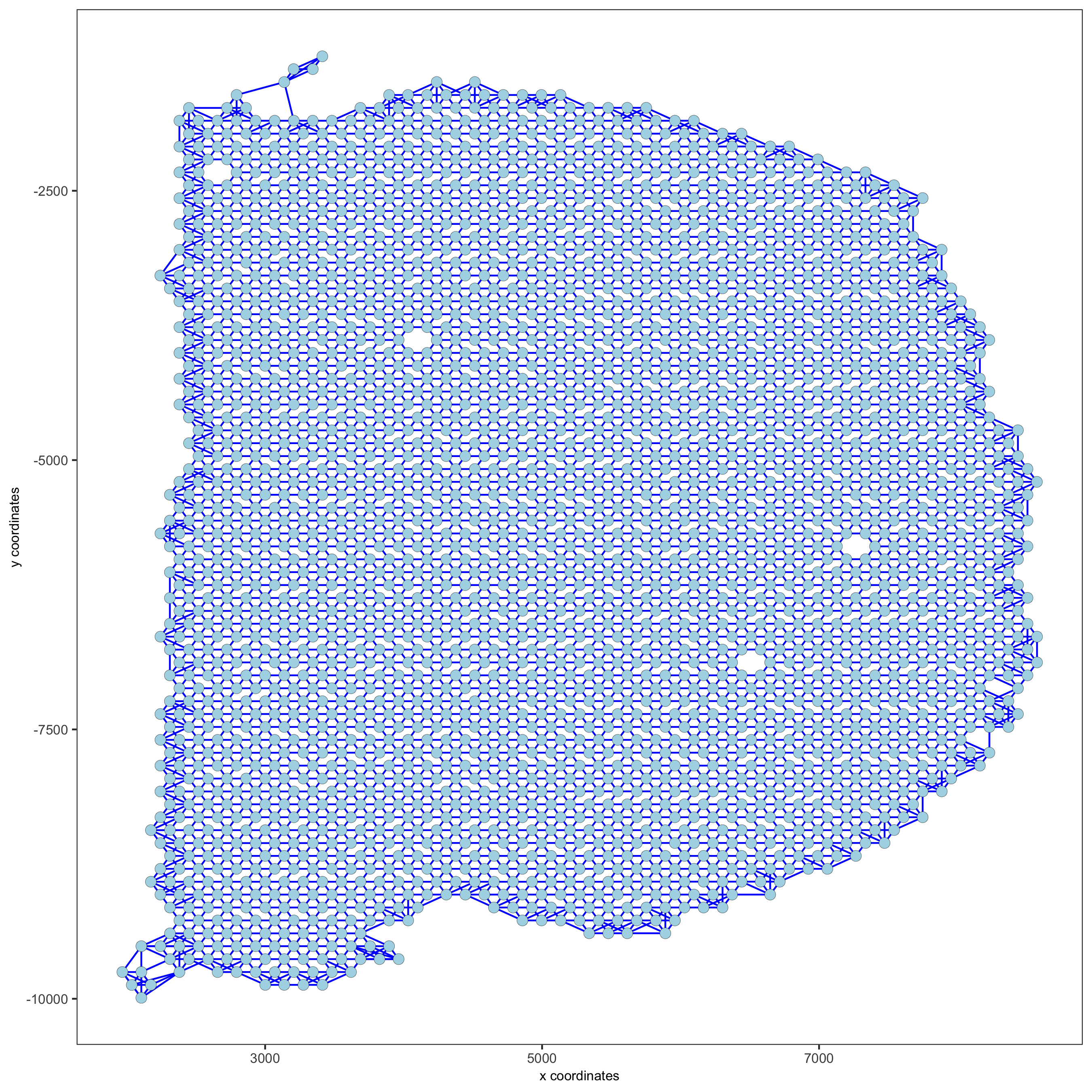
part 10: spatial genes
Spatial genes
## kmeans binarization
kmtest = binSpect(visium_brain, calc_hub = T, hub_min_int = 5,
spatial_network_name = 'spatial_network')
spatGenePlot(visium_brain, expression_values = 'scaled',
genes = kmtest$genes[1:6], cow_n_col = 2, point_size = 1.5,
save_param = list(save_name = '10_a_spatial_genes_km'))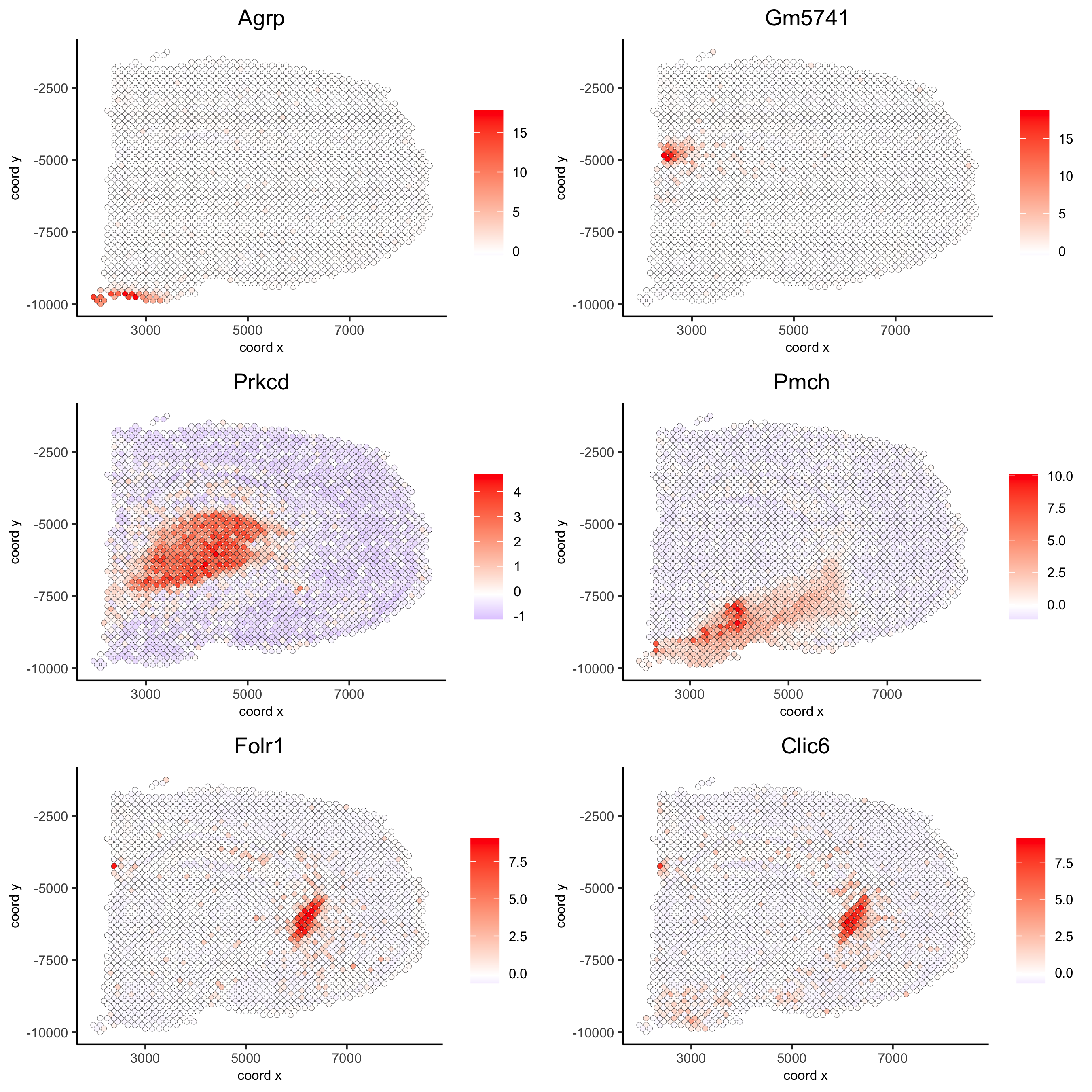
## rank binarization
ranktest = binSpect(visium_brain, bin_method = 'rank',
calc_hub = T, hub_min_int = 5,
spatial_network_name = 'spatial_network')
spatGenePlot(visium_brain, expression_values = 'scaled',
genes = ranktest$genes[1:6], cow_n_col = 2, point_size = 1.5,
save_param = list(save_name = '10_b_spatial_genes_rank'))![]()
Spatial patterns
# cluster the top 1500 spatial genes into 20 clusters
ext_spatial_genes = ranktest[1:1500,]$gene
# here we use existing detectSpatialCorGenes function to calculate pairwise distances between genes (but set network_smoothing=0 to use default clustering)
spat_cor_netw_DT = detectSpatialCorGenes(visium_brain,
method = 'network',
spatial_network_name = 'spatial_network',
subset_genes = ext_spatial_genes)
# cluster spatial genes
spat_cor_netw_DT = clusterSpatialCorGenes(spat_cor_netw_DT, name = 'spat_netw_clus', k = 20)
# visualize clusters
heatmSpatialCorGenes(visium_brain,
spatCorObject = spat_cor_netw_DT,
use_clus_name = 'spat_netw_clus',
heatmap_legend_param = list(title = NULL),
save_param = list(save_name="10_c_heatmap",
base_height = 6, base_width = 8, units = 'cm'))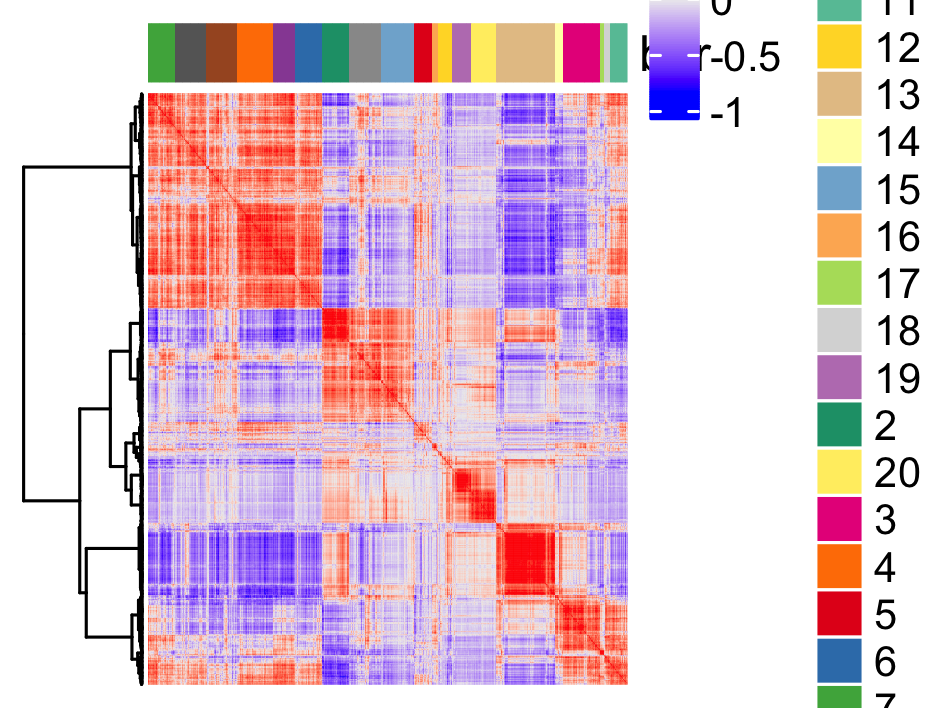
table(spat_cor_netw_DT$cor_clusters$spat_netw_clus)
coexpr_dt = data.table::data.table(genes = names(spat_cor_netw_DT$cor_clusters$spat_netw_clus),
cluster = spat_cor_netw_DT$cor_clusters$spat_netw_clus)
data.table::setorder(coexpr_dt, cluster)
top30_coexpr_dt = coexpr_dt[, head(.SD, 30) , by = cluster]
# do HMRF with different betas on 500 spatial genes
my_spatial_genes <- top30_coexpr_dt$genes
hmrf_folder = paste0(results_folder,'/','11_HMRF/')
if(!file.exists(hmrf_folder)) dir.create(hmrf_folder, recursive = T)
HMRF_spatial_genes = doHMRF(gobject = visium_brain,
expression_values = 'scaled',
spatial_genes = my_spatial_genes, k = 20,
spatial_network_name="spatial_network",
betas = c(0, 10, 5),
output_folder = paste0(hmrf_folder, '/', 'Spatial_genes/SG_topgenes_k20_scaled'))
visium_brain = addHMRF(gobject = visium_brain, HMRFoutput = HMRF_spatial_genes,
k = 20, betas_to_add = c(0, 10, 20, 30, 40),
hmrf_name = 'HMRF')
spatPlot(gobject = visium_brain, cell_color = 'HMRF_k20_b.40',
point_size = 2, save_param=c(save_name="10_d_spatPlot2D_HMRF"))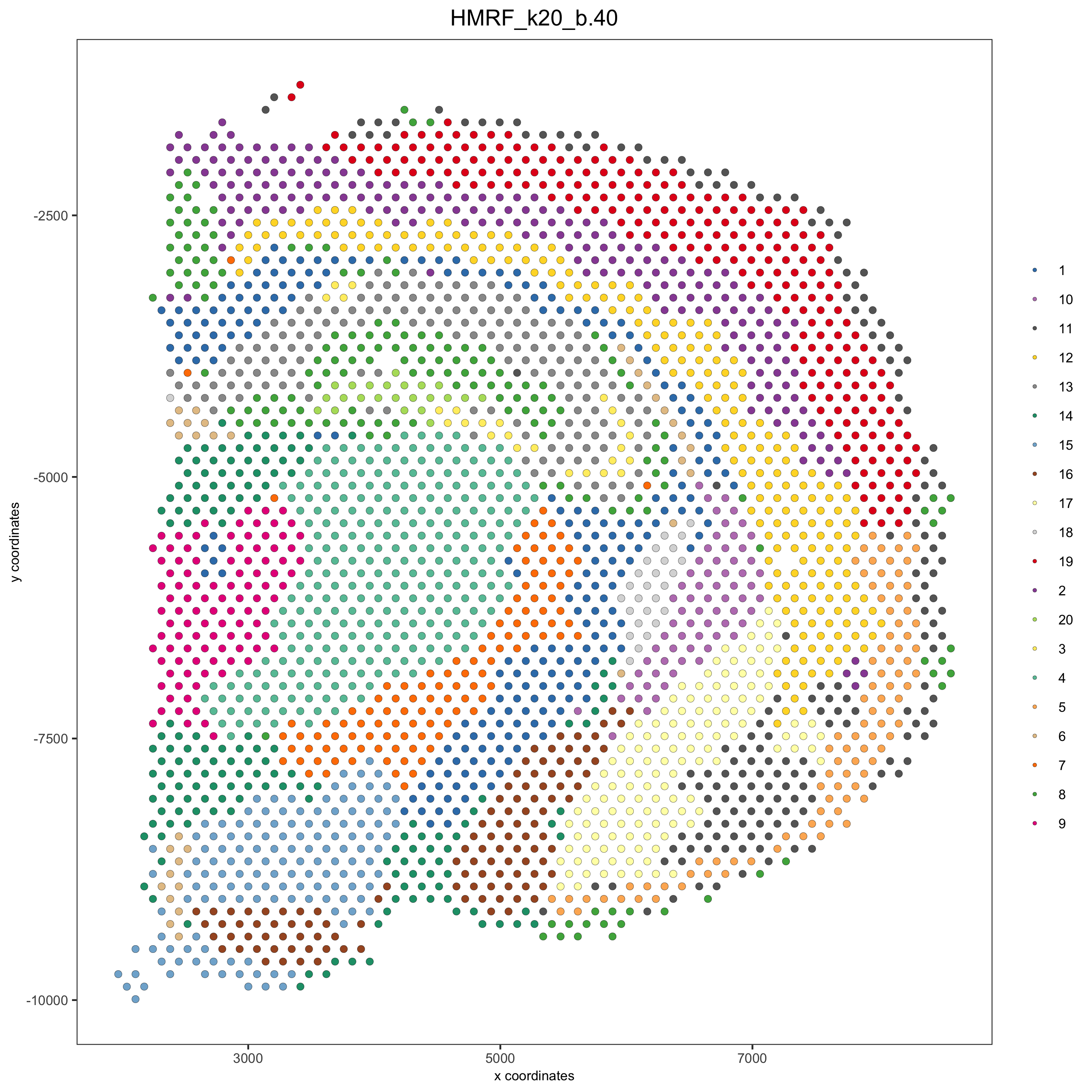
Export and create Giotto Viewer
# check which annotations are available
combineMetadata(visium_brain, spat_enr_names = 'PAGE')
# select annotations, reductions and expression values to view in Giotto Viewer
viewer_folder = paste0(results_folder, '/', 'mouse_Visium_brain_viewer')
exportGiottoViewer(gobject = visium_brain,
output_directory = viewer_folder,
spat_enr_names = 'PAGE',
factor_annotations = c('in_tissue',
'leiden_clus',
'HMRF_k20_b.40'),
numeric_annotations = c('nr_genes',
'clus_25'),
dim_reductions = c('tsne', 'umap'),
dim_reduction_names = c('tsne', 'umap'),
expression_values = 'scaled',
expression_rounding = 2,
overwrite_dir = T)