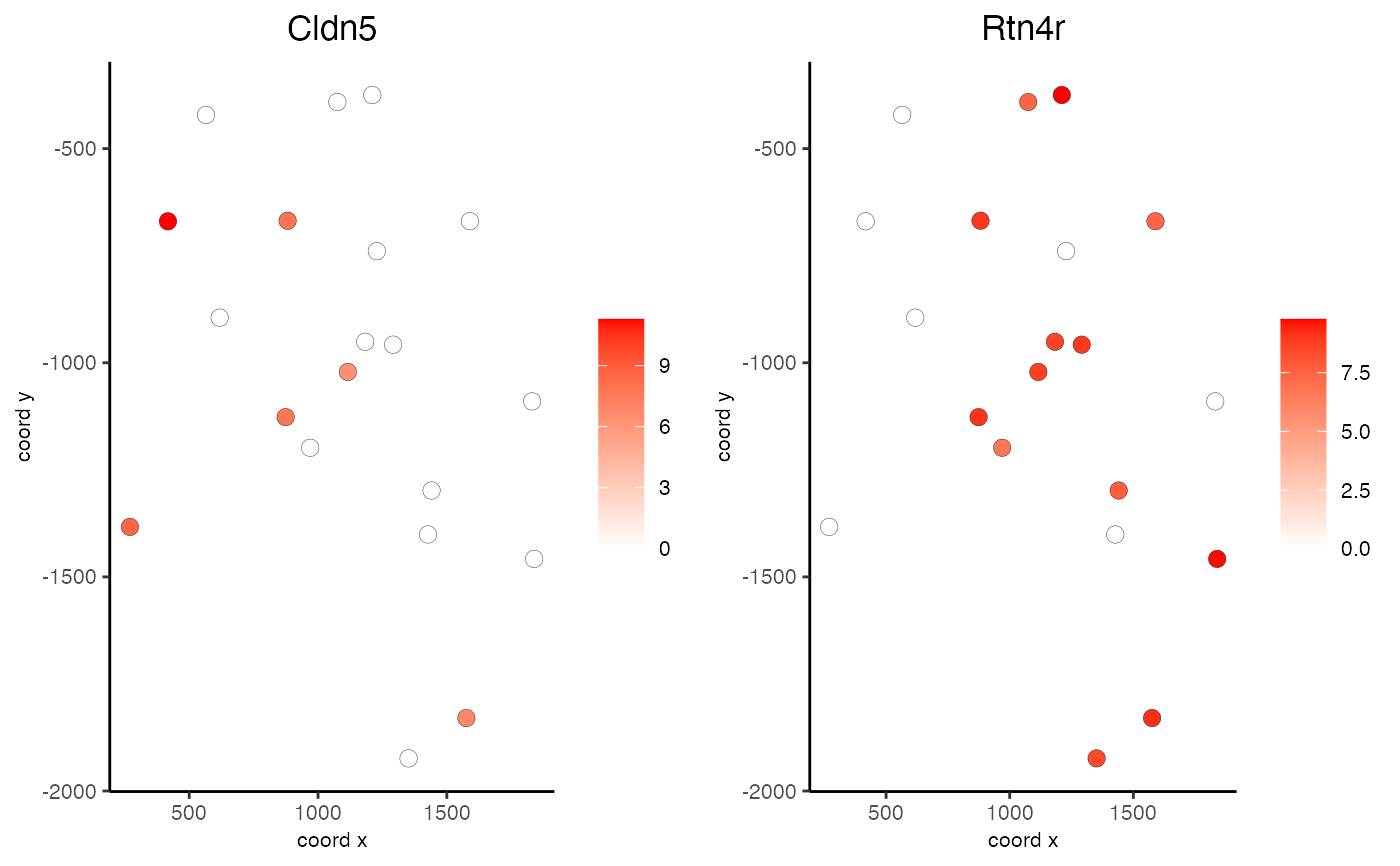
Visualize cells and gene expression according to spatial coordinates
spatGenePlot2D( gobject, show_image = F, gimage = NULL, image_name = "image", sdimx = "sdimx", sdimy = "sdimy", expression_values = c("normalized", "scaled", "custom"), genes, cell_color_gradient = c("blue", "white", "red"), gradient_midpoint = NULL, gradient_limits = NULL, show_network = F, network_color = NULL, spatial_network_name = "Delaunay_network", edge_alpha = NULL, show_grid = F, grid_color = NULL, spatial_grid_name = "spatial_grid", midpoint = 0, scale_alpha_with_expression = FALSE, point_shape = c("border", "no_border", "voronoi"), point_size = 1, point_alpha = 1, point_border_col = "black", point_border_stroke = 0.1, show_legend = T, legend_text = 8, background_color = "white", vor_border_color = "white", vor_alpha = 1, vor_max_radius = 200, axis_text = 8, axis_title = 8, cow_n_col = 2, cow_rel_h = 1, cow_rel_w = 1, cow_align = "h", show_plot = NA, return_plot = NA, save_plot = NA, save_param = list(), default_save_name = "spatGenePlot2D" )
Arguments
| gobject | giotto object |
|---|---|
| show_image | show a tissue background image |
| gimage | a giotto image |
| image_name | name of a giotto image |
| sdimx | x-axis dimension name (default = 'sdimx') |
| sdimy | y-axis dimension name (default = 'sdimy') |
| expression_values | gene expression values to use |
| genes | genes to show |
| cell_color_gradient | vector with 3 colors for numeric data |
| gradient_midpoint | midpoint for color gradient |
| gradient_limits | vector with lower and upper limits |
| show_network | show underlying spatial network |
| network_color | color of spatial network |
| spatial_network_name | name of spatial network to use |
| edge_alpha | alpha of edge |
| show_grid | show spatial grid |
| grid_color | color of spatial grid |
| spatial_grid_name | name of spatial grid to use |
| midpoint | expression midpoint |
| scale_alpha_with_expression | scale expression with ggplot alpha parameter |
| point_shape | shape of points (border, no_border or voronoi) |
| point_size | size of point (cell) |
| point_alpha | transparancy of points |
| point_border_col | color of border around points |
| point_border_stroke | stroke size of border around points |
| show_legend | show legend |
| legend_text | size of legend text |
| background_color | color of plot background |
| vor_border_color | border colorr for voronoi plot |
| vor_alpha | transparancy of voronoi 'cells' |
| vor_max_radius | maximum radius for voronoi 'cells' |
| axis_text | size of axis text |
| axis_title | size of axis title |
| cow_n_col | cowplot param: how many columns |
| cow_rel_h | cowplot param: relative height |
| cow_rel_w | cowplot param: relative width |
| cow_align | cowplot param: how to align |
| show_plot | show plots |
| return_plot | return ggplot object |
| save_plot | directly save the plot [boolean] |
| save_param | list of saving parameters, see |
| default_save_name | default save name for saving, don't change, change save_name in save_param |
Value
ggplot
Details
Description of parameters.
See also
Other spatial gene expression visualizations:
spatGenePlot3D(),
spatGenePlot()
Examples
data(mini_giotto_single_cell) all_genes = slot(mini_giotto_single_cell, 'gene_ID') selected_genes = all_genes[1:2] spatGenePlot2D(mini_giotto_single_cell, genes = selected_genes, point_size = 3)