osmFISH mouse SS cortex
Source:vignettes/mouse_osmFISH_SScortex_200915.Rmd
mouse_osmFISH_SScortex_200915.Rmd#> Warning: This tutorial was written with Giotto version 0.3.6.9042, your version
#> is 1.0.3.This is a more recent version and results should be reproduciblelibrary(Giotto) # 1. set working directory results_folder = '/path/to/directory/' # 2. set giotto python path # set python path to your preferred python version path # set python path to NULL if you want to automatically install (only the 1st time) and use the giotto miniconda environment python_path = NULL if(is.null(python_path)) { installGiottoEnvironment() }
Dataset explanation
Codeluppi et al. created a cyclic single-molecule fluorescence in situ hybridization (osmFISH) technology and define the cellular organization of the somatosensory cortex with the expression of 33 genes in 5,328 cells.
Dataset download
The osmFISH data to run this tutorial can be found here. Alternatively you can use the getSpatialDataset to automatically download this dataset like we do in this example.
# download data to working directory #### # if wget is installed, set method = 'wget' getSpatialDataset(dataset = 'osmfish_SS_cortex', directory = results_folder, method = 'wget')
Part 1: Giotto global instructions and preparations
## instructions allow us to automatically save all plots into a chosen results folder instrs = createGiottoInstructions(save_plot = TRUE, show_plot = FALSE, save_dir = results_folder, python_path = python_path) expr_path = paste0(results_folder, "osmFISH_prep_expression.txt") loc_path = paste0(results_folder, "osmFISH_prep_cell_coordinates.txt") meta_path = paste0(results_folder, "osmFISH_prep_cell_metadata.txt")
part 2: Create Giotto object & process data
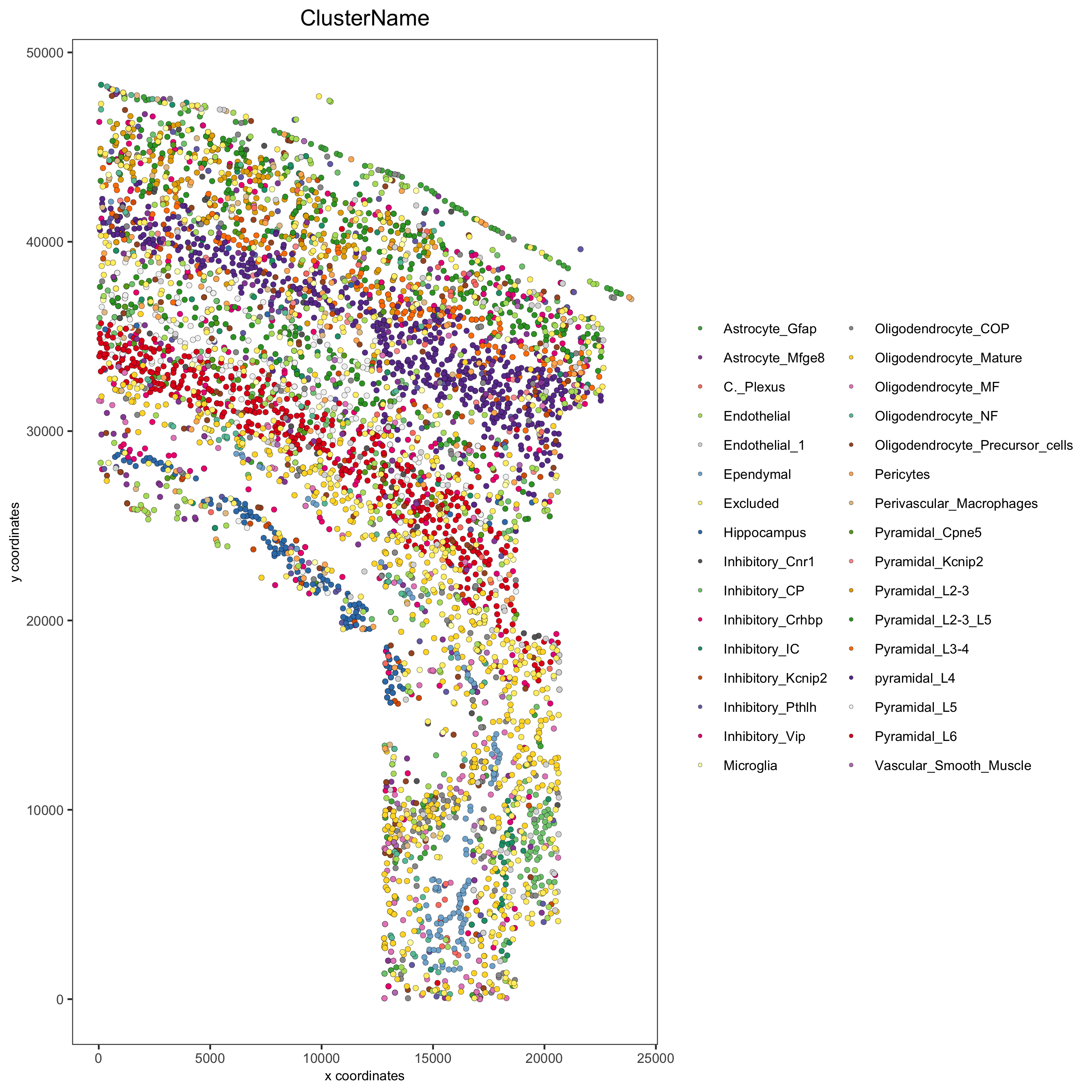
## create osm_test <- createGiottoObject(raw_exprs = expr_path, spatial_locs = loc_path, instructions = instrs) showGiottoInstructions(osm_test) ## add field annotation metadata = data.table::fread(file = meta_path) osm_test = addCellMetadata(osm_test, new_metadata = metadata, by_column = T, column_cell_ID = 'CellID') ## filter osm_test <- filterGiotto(gobject = osm_test, expression_threshold = 1, gene_det_in_min_cells = 10, min_det_genes_per_cell = 10, expression_values = c('raw'), verbose = T) ## normalize # 1. standard z-score way osm_test <- normalizeGiotto(gobject = osm_test) # 2. osmFISH way raw_expr_matrix = osm_test@raw_exprs norm_genes = (raw_expr_matrix/rowSums_giotto(raw_expr_matrix)) * nrow(raw_expr_matrix) norm_genes_cells = t_giotto((t_giotto(norm_genes)/colSums_giotto(norm_genes)) * ncol(raw_expr_matrix)) osm_test@custom_expr = norm_genes_cells ## add gene & cell statistics osm_test <- addStatistics(gobject = osm_test) ## add gene & cell statistics osm_test <- addStatistics(gobject = osm_test) # save according to giotto instructions spatPlot(gobject = osm_test, cell_color = 'ClusterName', point_size = 1.5, save_param = list(save_name = '2_a_original_clusters'))
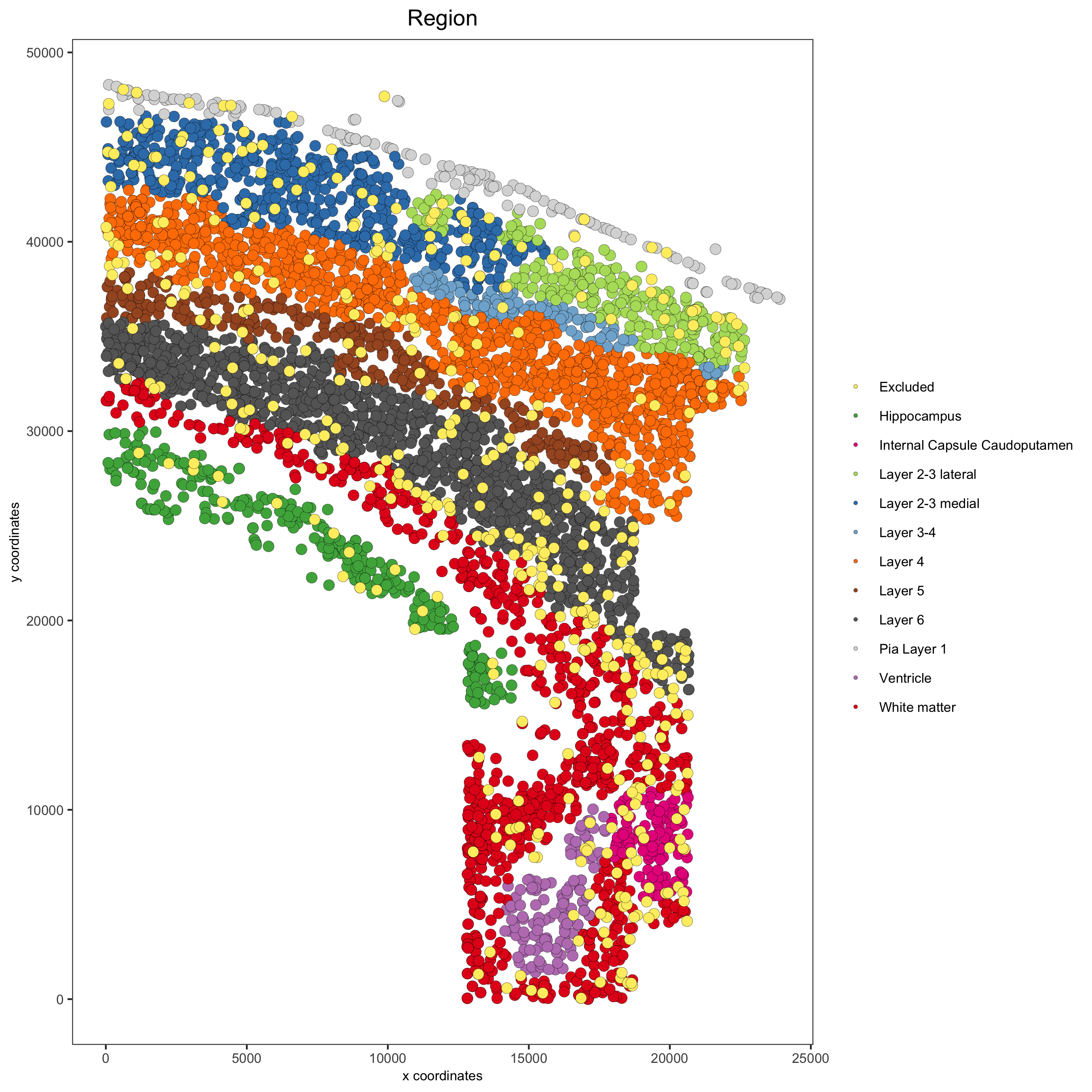
spatPlot(gobject = osm_test, cell_color = 'Region', save_param = list(save_name = '2_b_original_regions'))
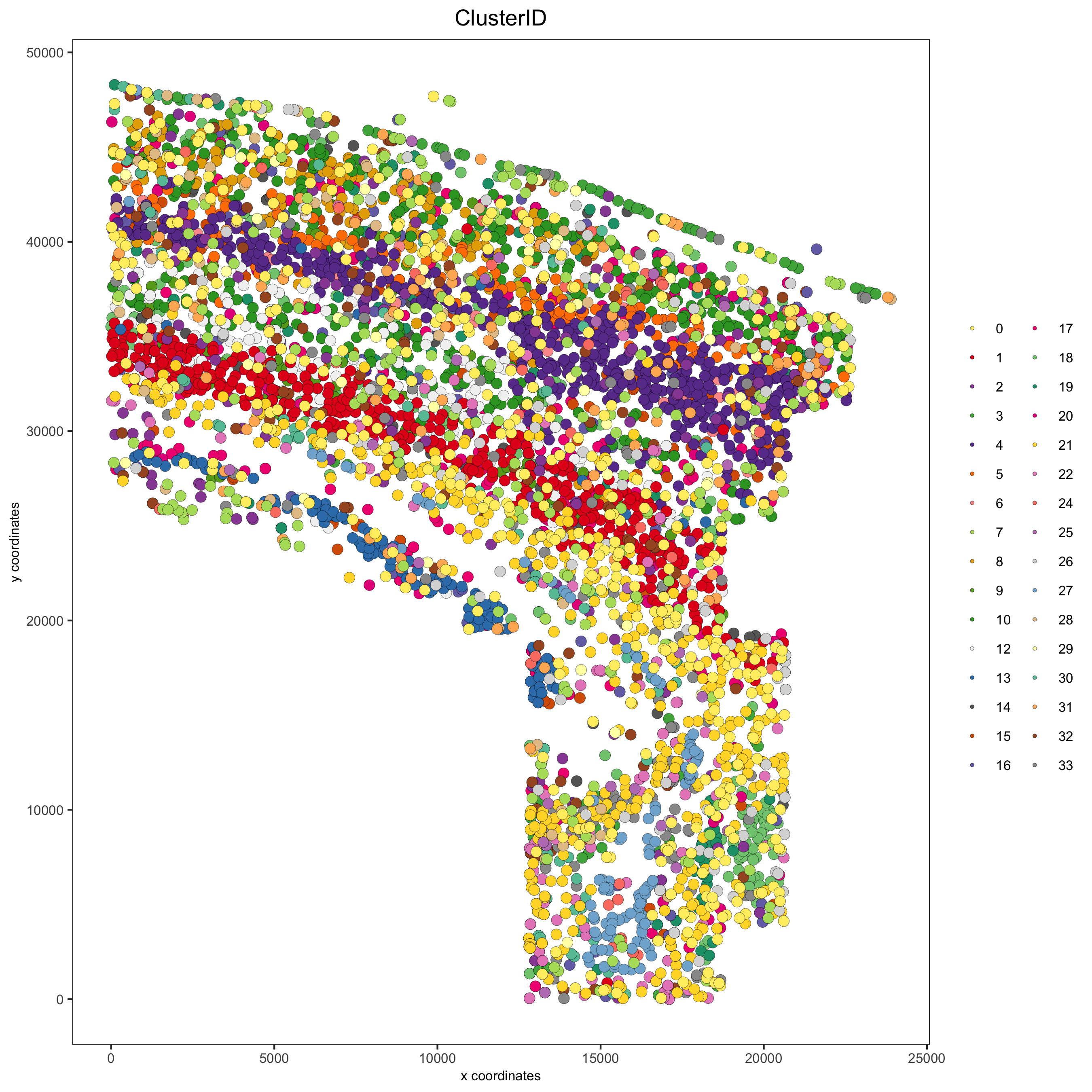
spatPlot(gobject = osm_test, cell_color = 'ClusterID', save_param = list(save_name = '2_c_clusterID'))
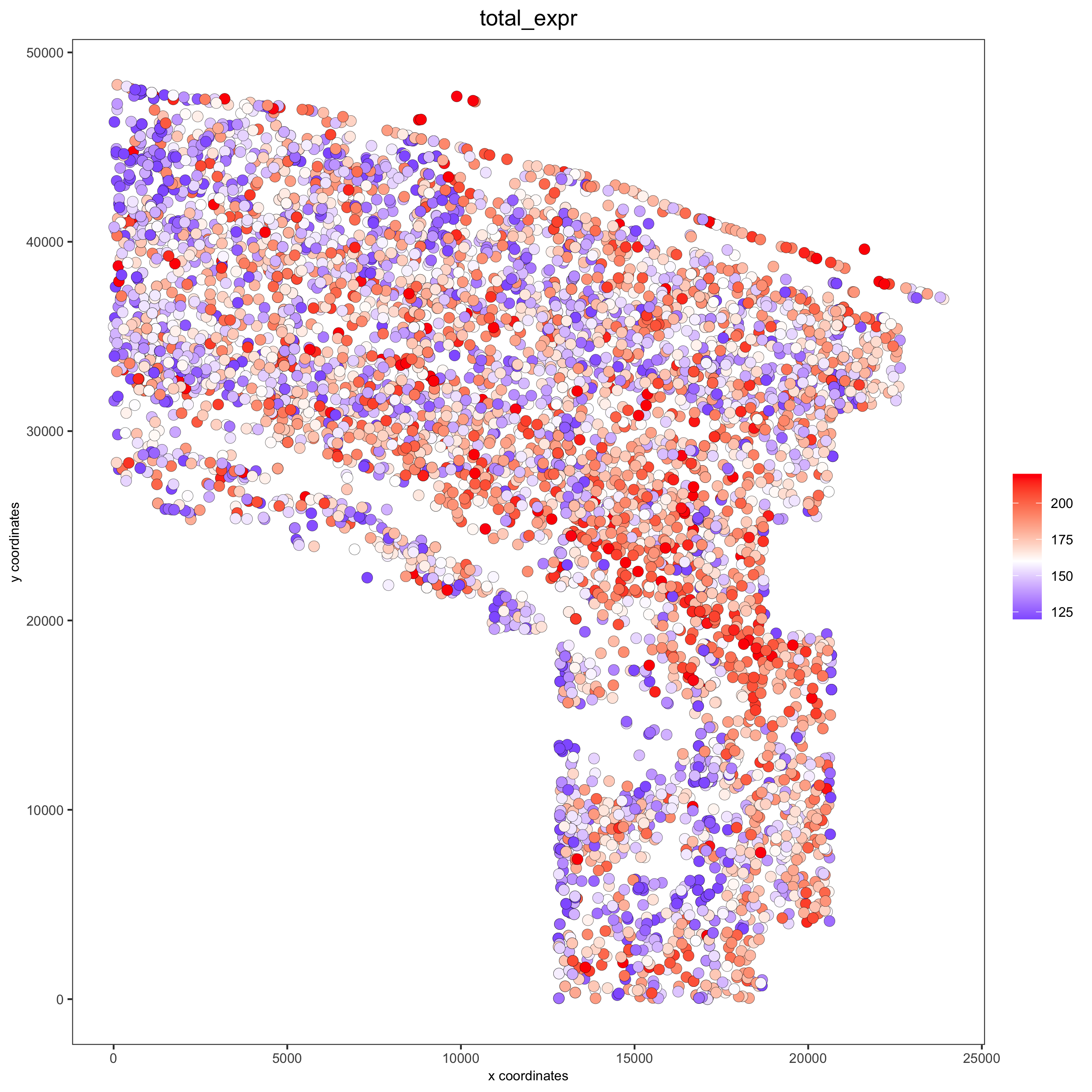
spatPlot(gobject = osm_test, cell_color = 'total_expr', color_as_factor = F, gradient_midpoint = 160, gradient_limits = c(120,220), save_param = list(save_name = '2_d_total_expr_limits'))
Part 3: Dimension reduction
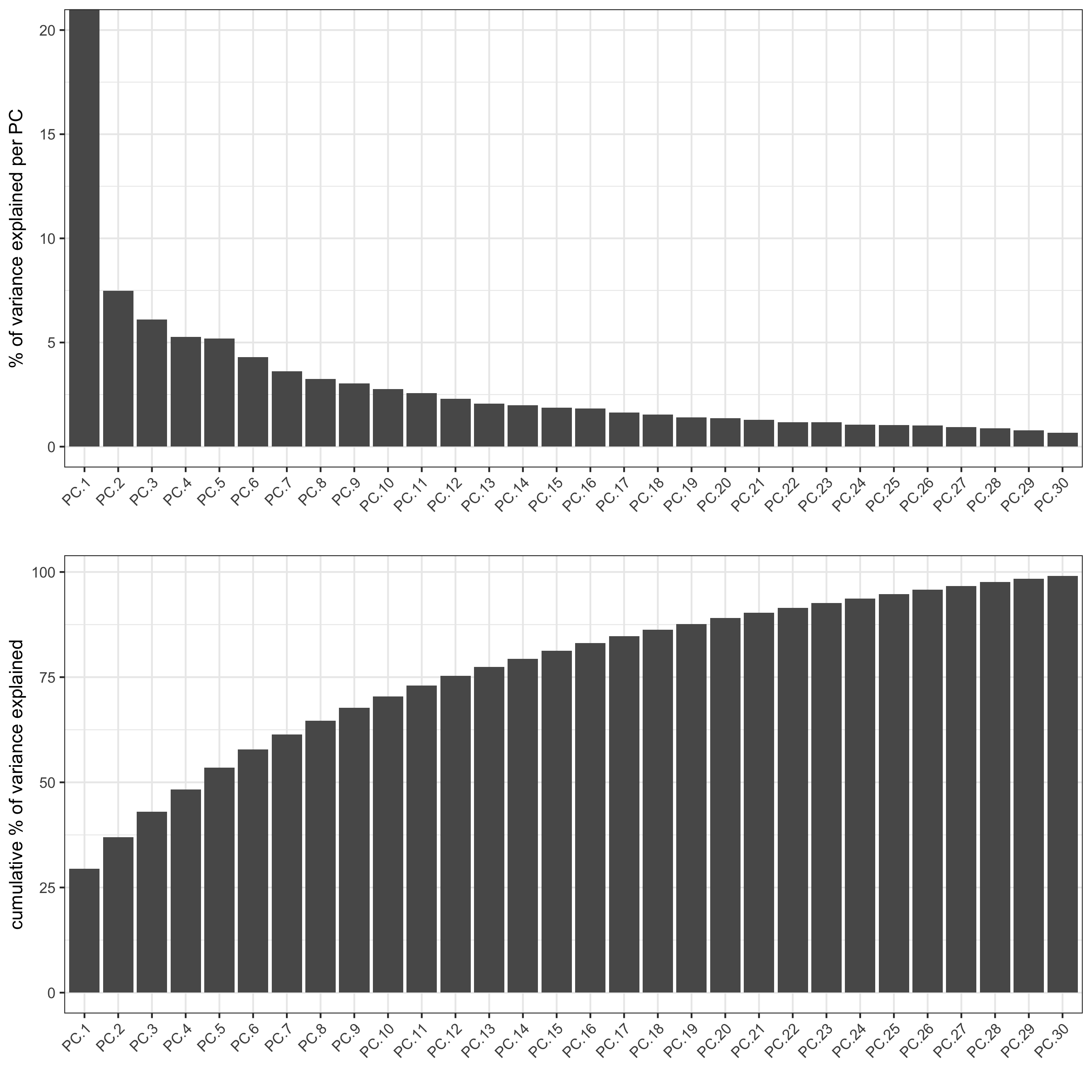
## highly variable genes (HVG) # only 33 genes so use all genes ## run PCA on expression values (default) osm_test <- runPCA(gobject = osm_test, expression_values = 'custom', scale_unit = F, center = F) screePlot(osm_test, ncp = 30, save_param = list(save_name = '3_a_screeplot'))
## run UMAP and tSNE on PCA space (default) osm_test <- runUMAP(osm_test, dimensions_to_use = 1:31, n_threads = 4) plotUMAP(gobject = osm_test, save_param = list(save_name = '3_c_UMAP_reduction.png'))
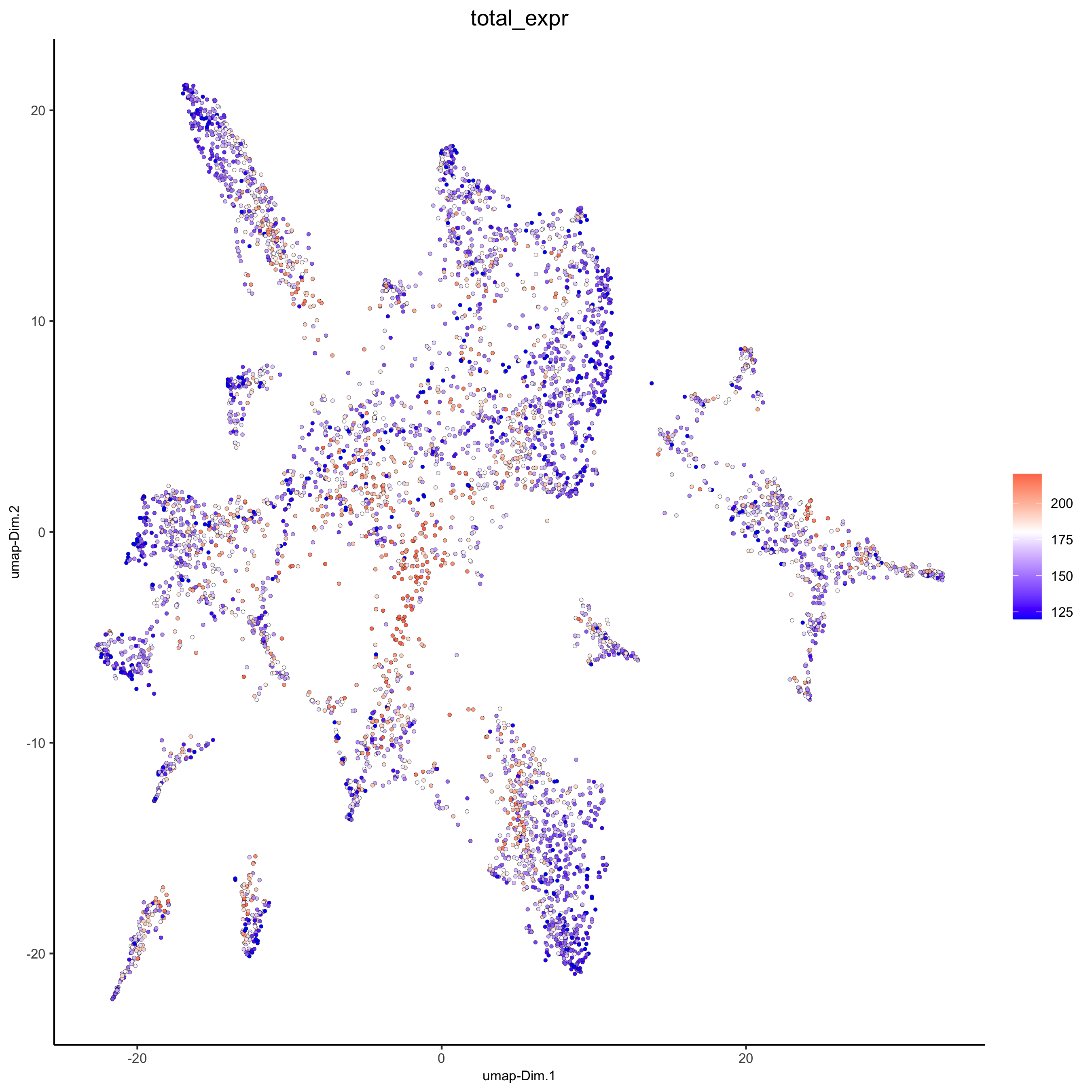
plotUMAP(gobject = osm_test, cell_color = 'total_expr', color_as_factor = F, gradient_midpoint = 180, gradient_limits = c(120, 220), save_param = list(save_name = '3_d_UMAP_reduction_expression.png'))
osm_test <- runtSNE(osm_test, dimensions_to_use = 1:31, perplexity = 70, check_duplicates = F) plotTSNE(gobject = osm_test, save_param = list(save_name = '3_e_tSNE_reduction'))
Part 4: Cluster
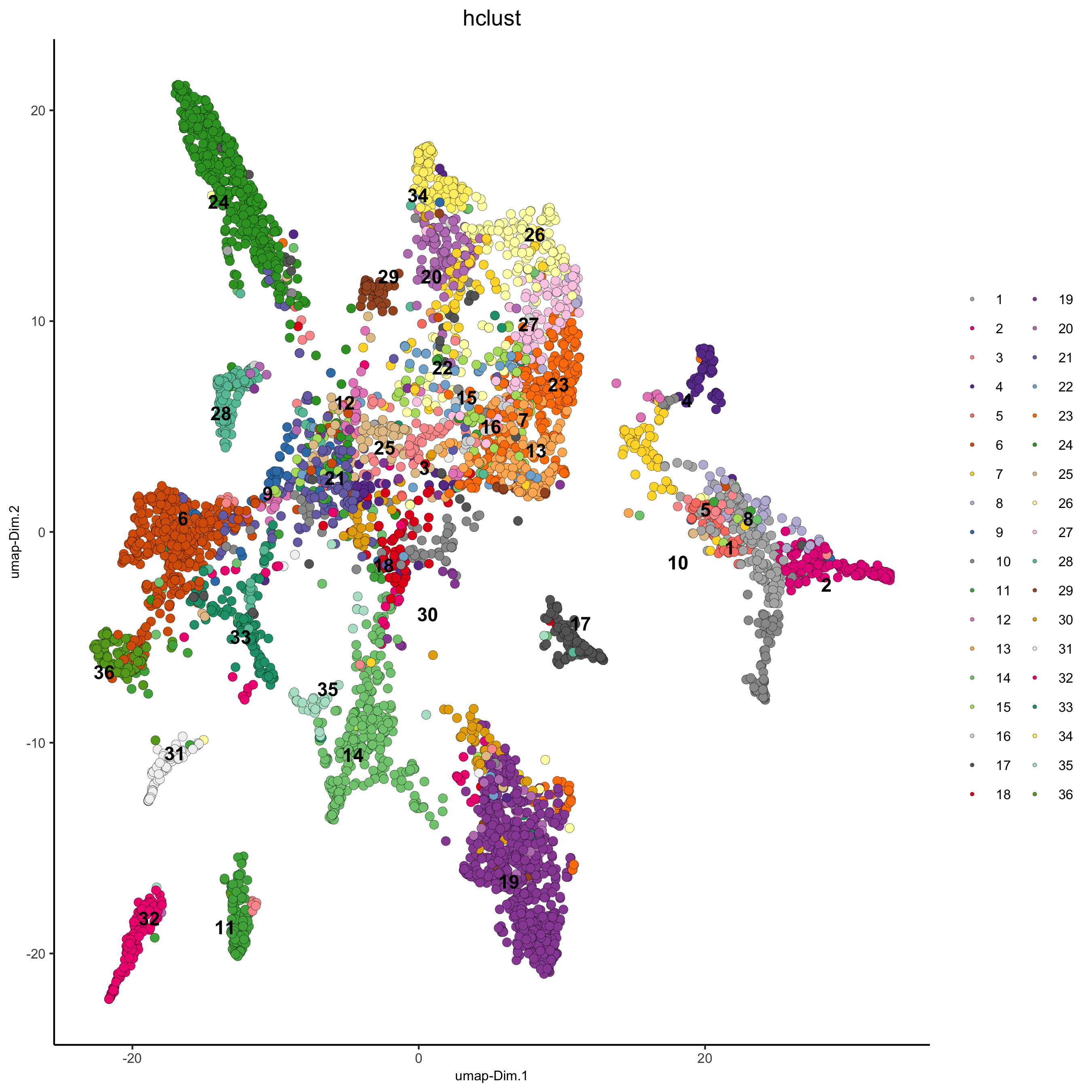
## hierarchical clustering osm_test = doHclust(gobject = osm_test, expression_values = 'custom', k = 36) plotUMAP(gobject = osm_test, cell_color = 'hclust', point_size = 2.5, show_NN_network = F, edge_alpha = 0.05, save_param = list(save_name = '4_a_UMAP_hclust'))
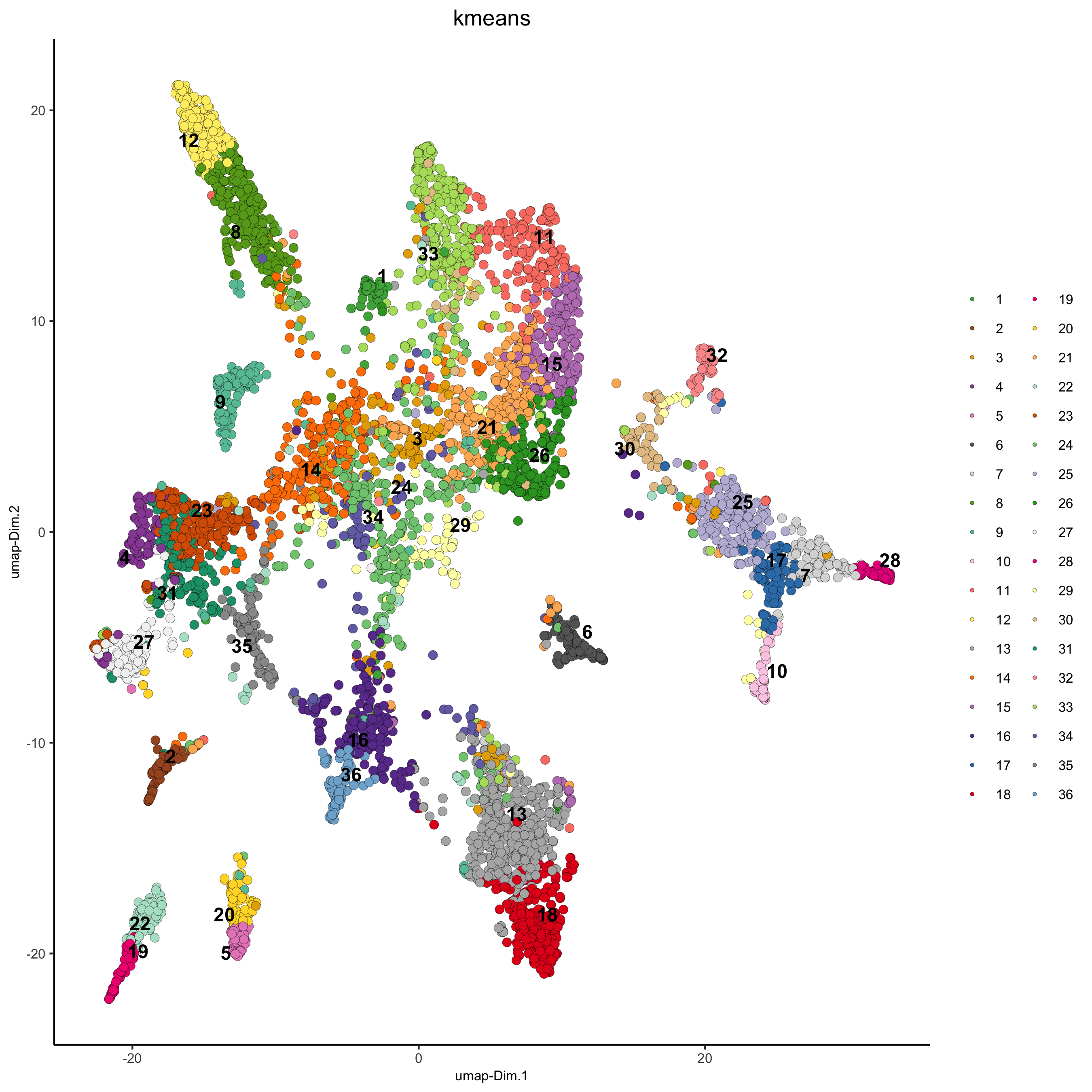
## kmeans clustering osm_test = doKmeans(gobject = osm_test, dim_reduction_to_use = 'pca', dimensions_to_use = 1:20, centers = 36, nstart = 2000) plotUMAP(gobject = osm_test, cell_color = 'kmeans', point_size = 2.5, show_NN_network = F, edge_alpha = 0.05, save_param = list(save_name = '4_b_UMAP_kmeans'))
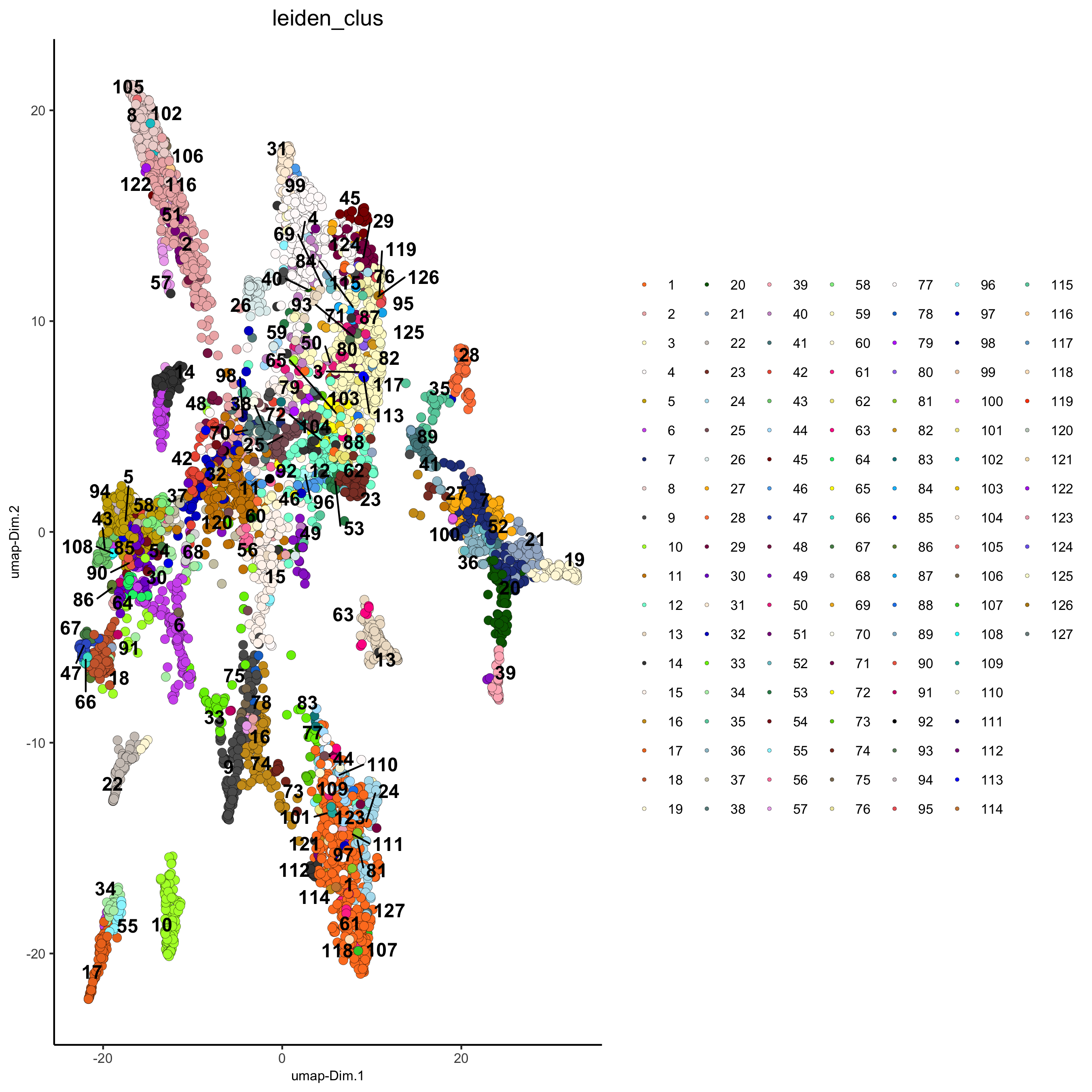
## Leiden clustering strategy: # 1. overcluster # 2. merge small clusters that are highly similar # sNN network (default) osm_test <- createNearestNetwork(gobject = osm_test, dimensions_to_use = 1:31, k = 12) osm_test <- doLeidenCluster(gobject = osm_test, resolution = 0.09, n_iterations = 1000) plotUMAP(gobject = osm_test, cell_color = 'leiden_clus', point_size = 2.5, show_NN_network = F, edge_alpha = 0.05, save_param = list(save_name = '4_c_UMAP_leiden'))
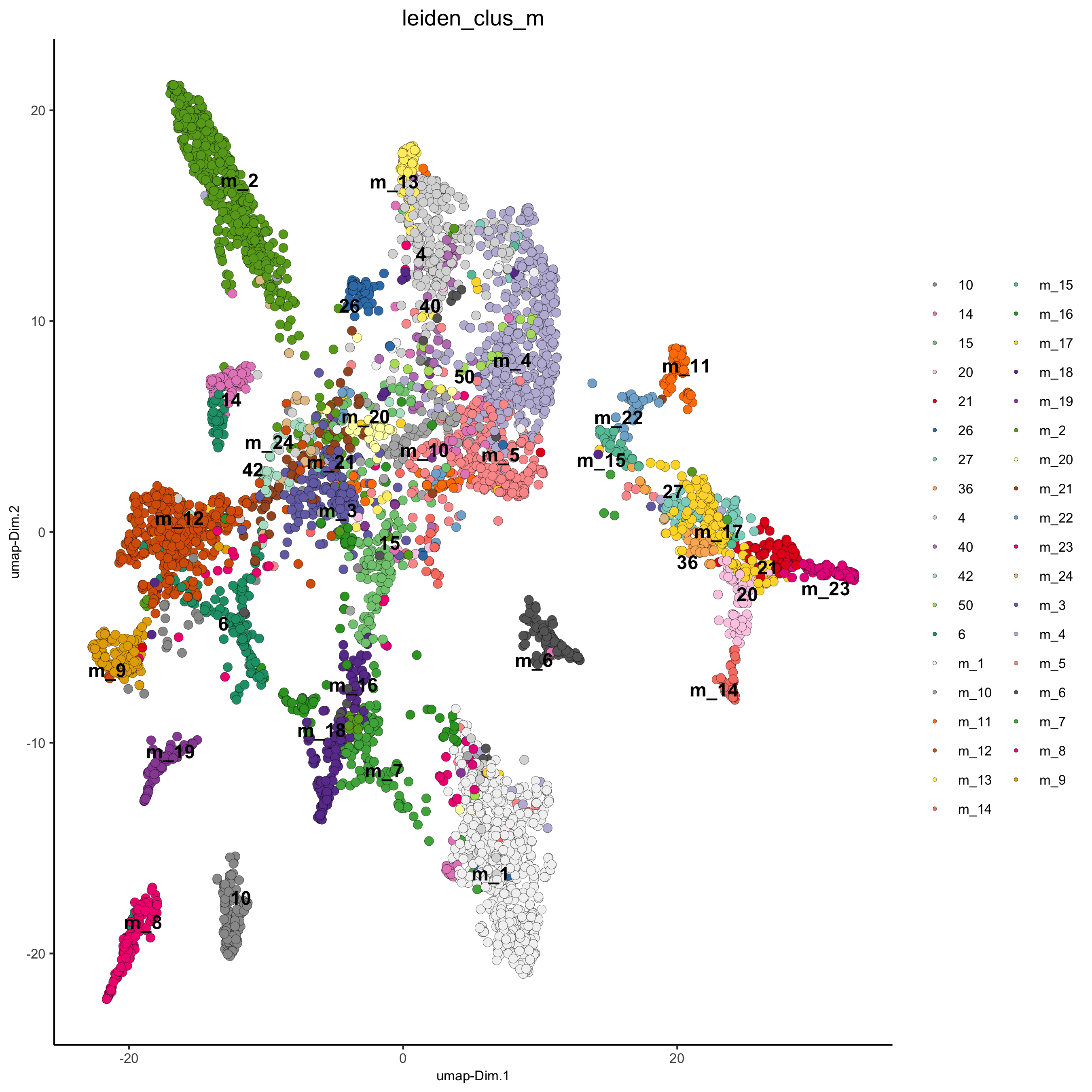
# merge small groups based on similarity leiden_similarities = getClusterSimilarity(osm_test, expression_values = 'custom', cluster_column = 'leiden_clus') osm_test = mergeClusters(osm_test, expression_values = 'custom', cluster_column = 'leiden_clus', new_cluster_name = 'leiden_clus_m', max_group_size = 30, force_min_group_size = 25, max_sim_clusters = 10, min_cor_score = 0.7) plotUMAP(gobject = osm_test, cell_color = 'leiden_clus_m', point_size = 2.5, show_NN_network = F, edge_alpha = 0.05, save_param = list(save_name = '4_d_UMAP_leiden_merged'))
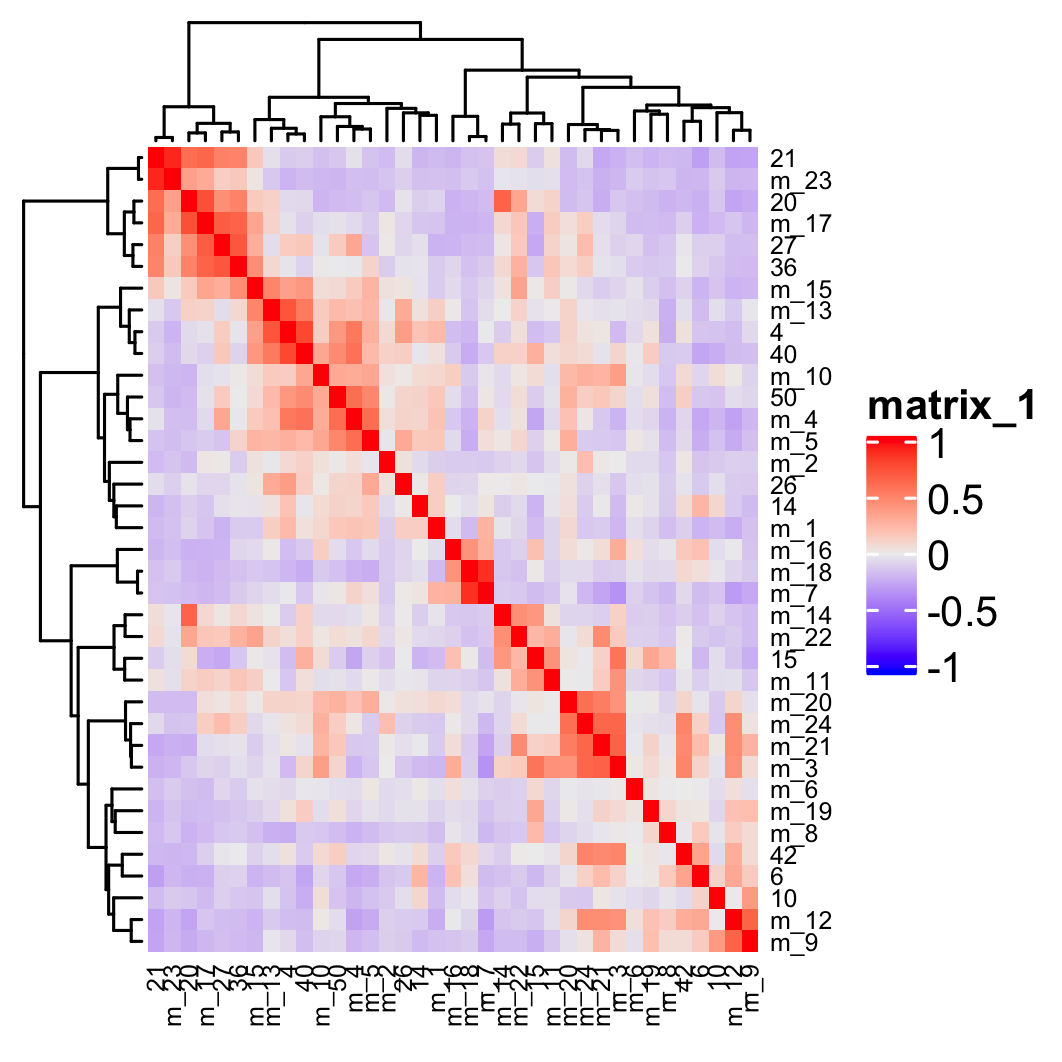
## show cluster relationships showClusterHeatmap(gobject = osm_test, expression_values = 'custom', cluster_column = 'leiden_clus_m', save_param = list(save_name = '4_e_heatmap', units = 'cm'), row_names_gp = grid::gpar(fontsize = 6), column_names_gp = grid::gpar(fontsize = 6))
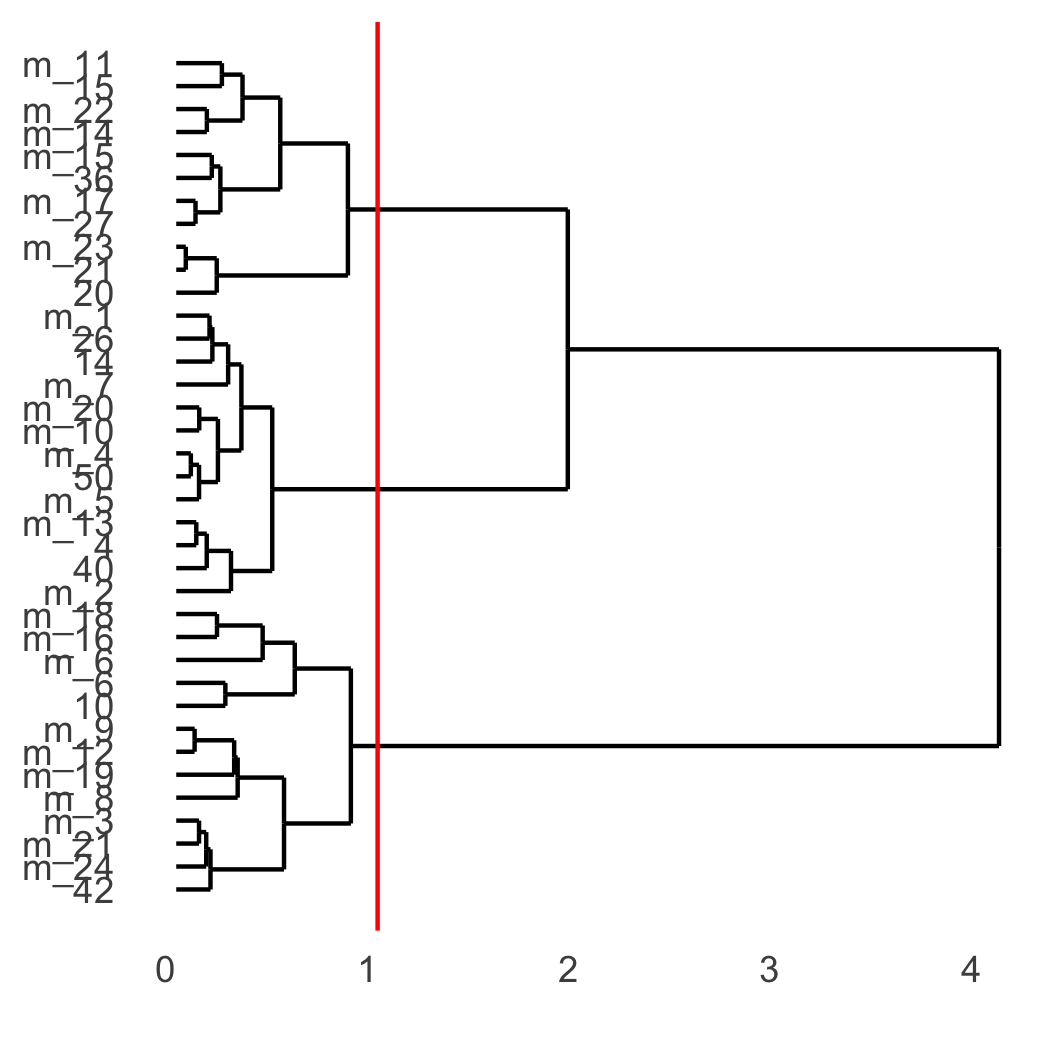
showClusterDendrogram(osm_test, cluster_column = 'leiden_clus_m', h = 1, rotate = T, save_param = list(save_name = '4_f_dendro', units = 'cm'))
Part 5: Co-visualize
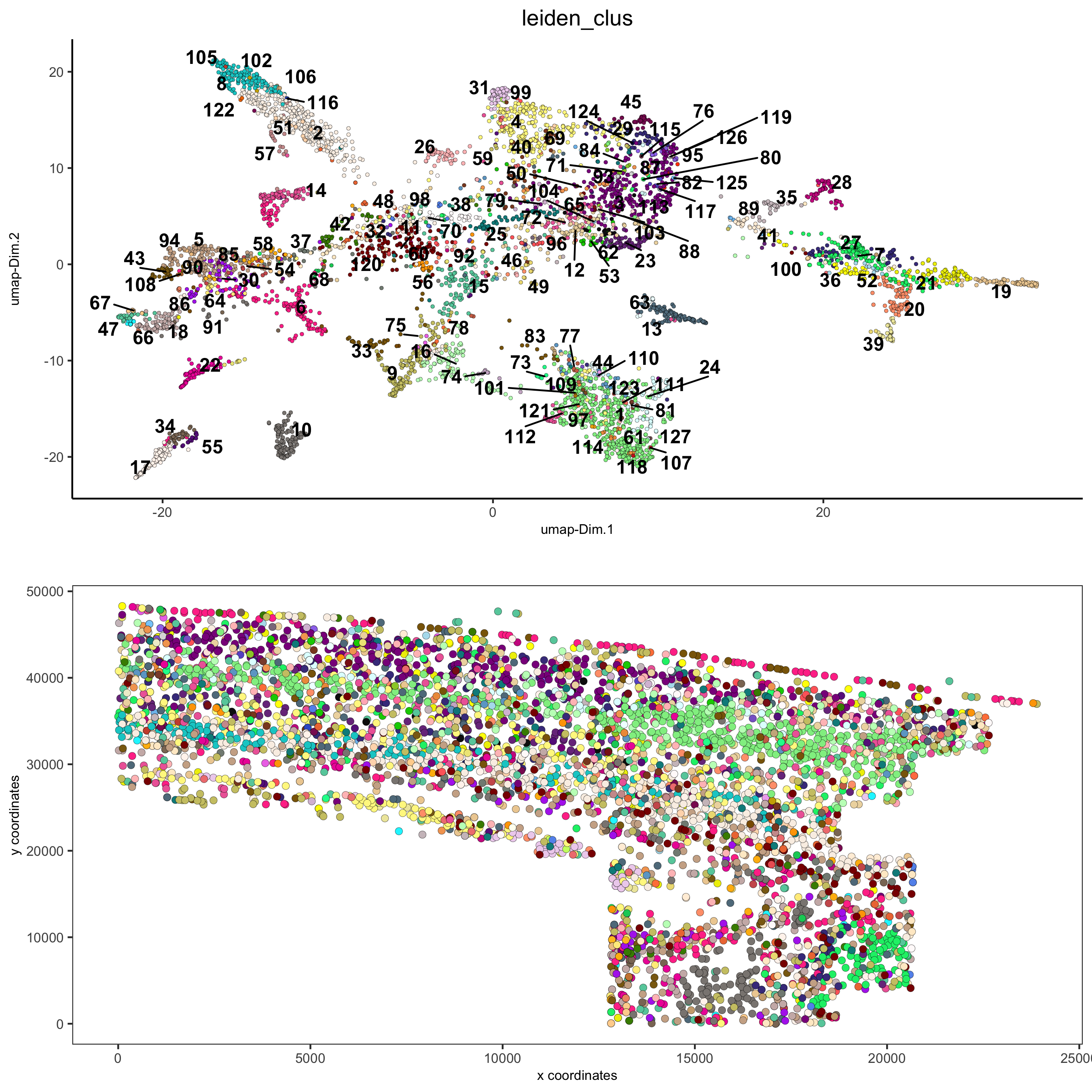
# expression and spatial spatDimPlot2D(gobject = osm_test, cell_color = 'leiden_clus', spat_point_size = 2, save_param = list(save_name = '5_a_covis_leiden'))
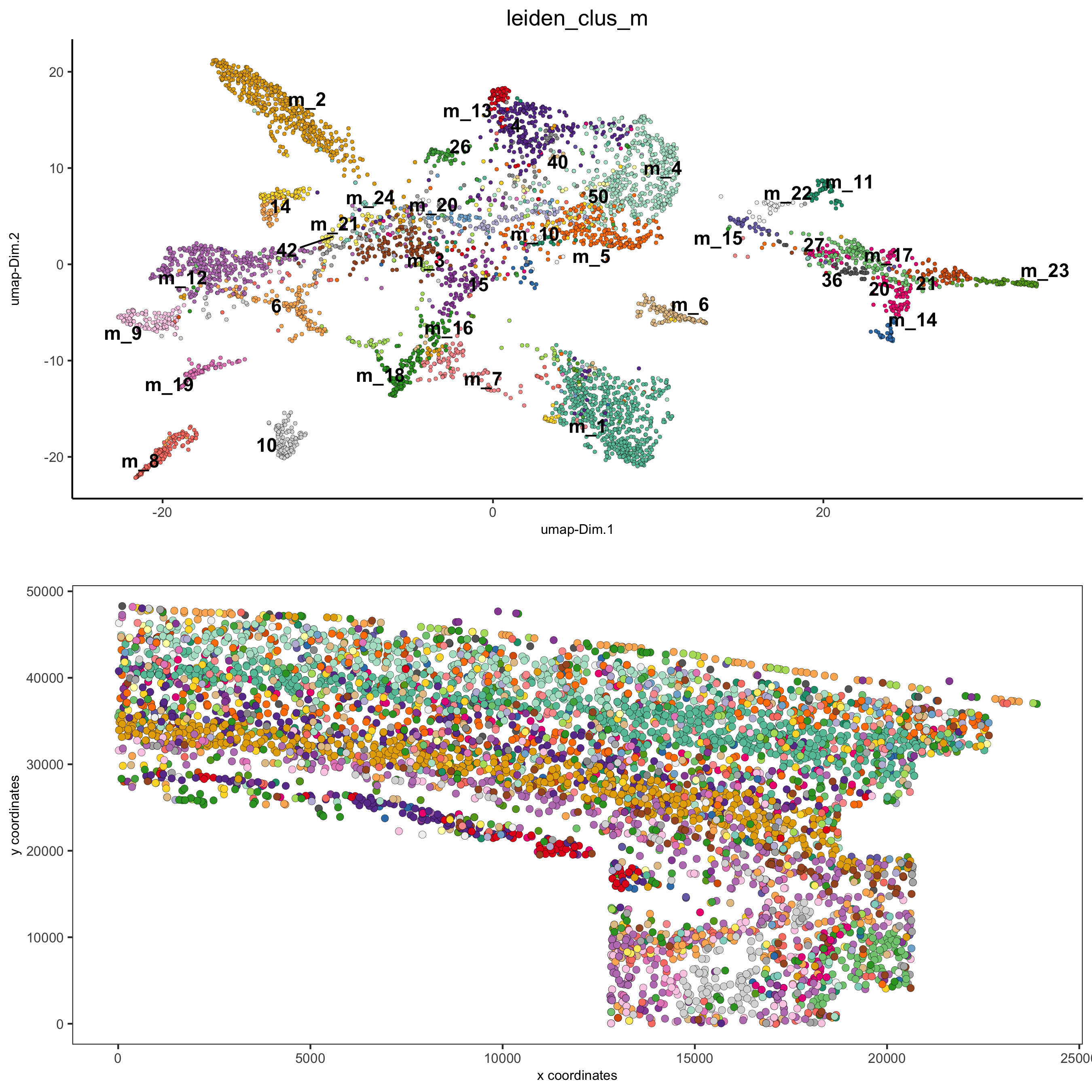
spatDimPlot2D(gobject = osm_test, cell_color = 'leiden_clus_m', spat_point_size = 2, save_param = list(save_name = '5_b_covis_leiden_m'))
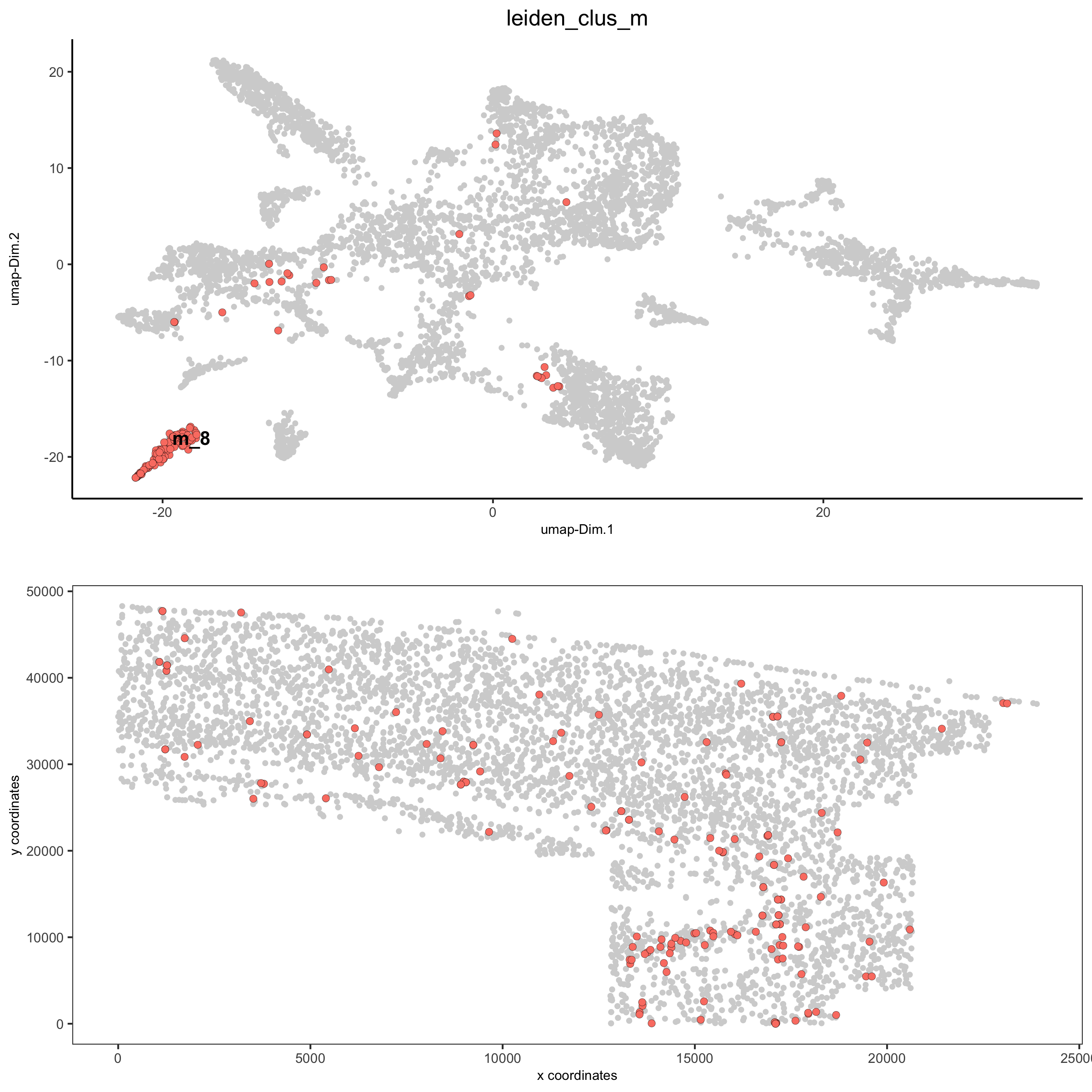
spatDimPlot2D(gobject = osm_test, cell_color = 'leiden_clus_m', dim_point_size = 2, spat_point_size = 2, select_cell_groups = 'm_8', save_param = list(save_name = '5_c_covis_leiden_merged_selected'))
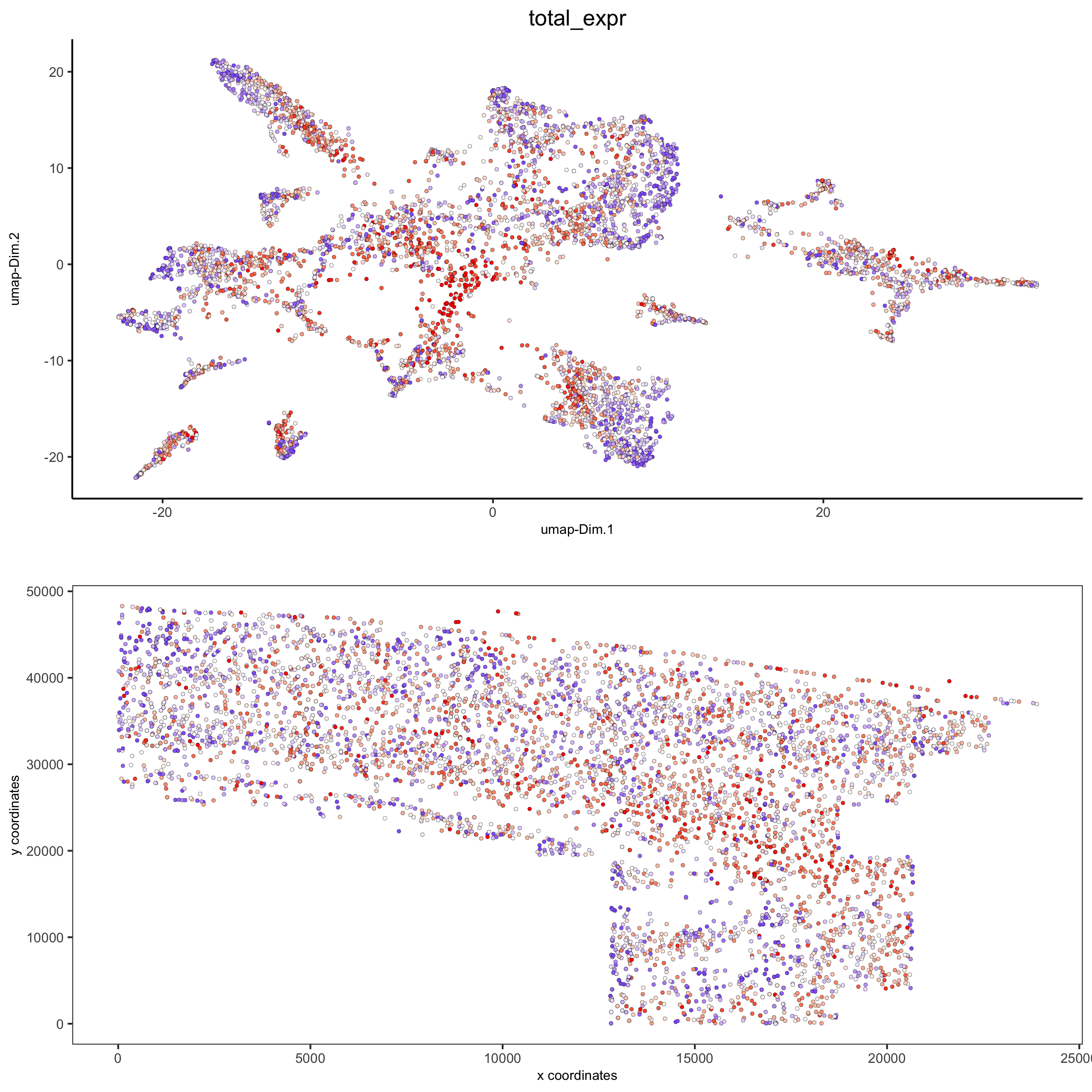
spatDimPlot2D(gobject = osm_test, cell_color = 'total_expr', color_as_factor = F, gradient_midpoint = 160, gradient_limits = c(120,220), save_param = list(save_name = '5_d_total_expr'))
Part 6: Differential expression
## split dendrogram nodes ## dendsplits = getDendrogramSplits(gobject = osm_test, expression_values = 'custom', cluster_column = 'leiden_clus_m') split_3_markers = findGiniMarkers(gobject = osm_test, expression_values = 'custom', cluster_column = 'leiden_clus_m', group_1 = unlist(dendsplits[3]$tree_1), group_2 = unlist(dendsplits[3]$tree_2))
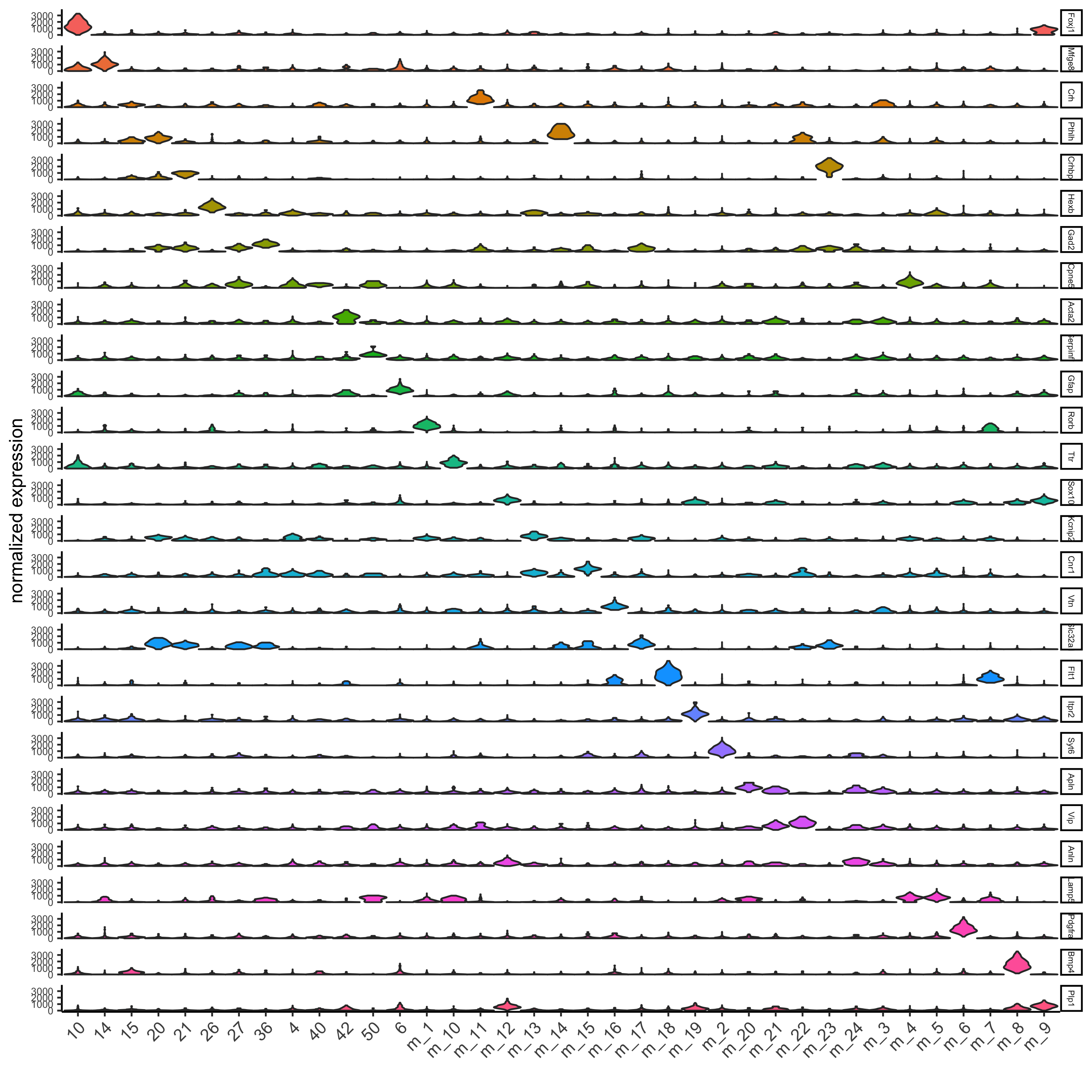
## Individual populations ## markers = findMarkers_one_vs_all(gobject = osm_test, method = 'scran', expression_values = 'custom', cluster_column = 'leiden_clus_m', min_genes = 2, rank_score = 2) ## violinplot topgenes = markers[, head(.SD, 1), by = 'cluster']$genes violinPlot(osm_test, genes = unique(topgenes), cluster_column = 'leiden_clus_m', expression_values = 'custom', strip_text = 5, strip_position = 'right', save_param = c(save_name = '6_a_violinplot'))
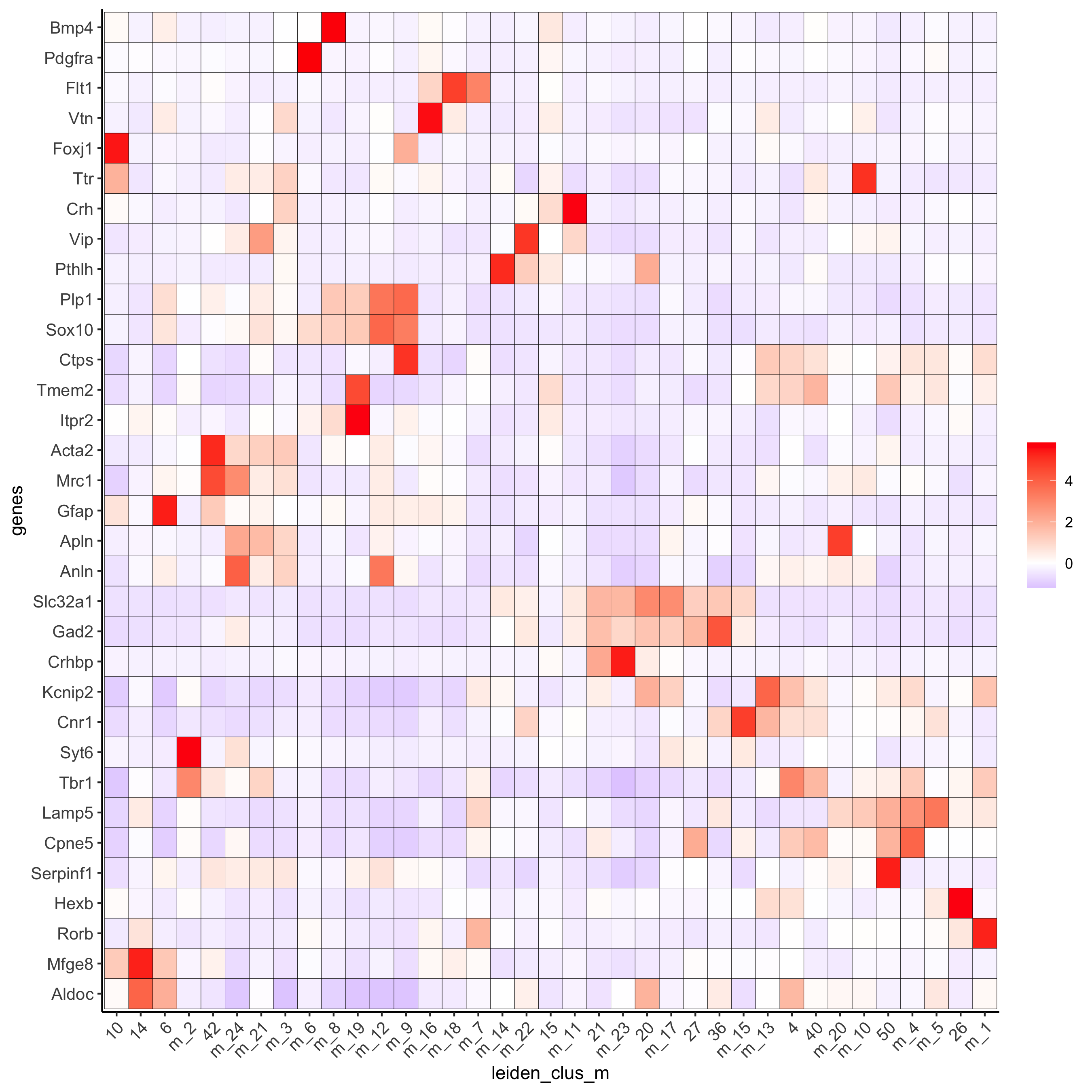
plotMetaDataHeatmap(osm_test, expression_values = 'custom', metadata_cols = c('leiden_clus_m'), save_param = c(save_name = '6_b_metaheatmap'))
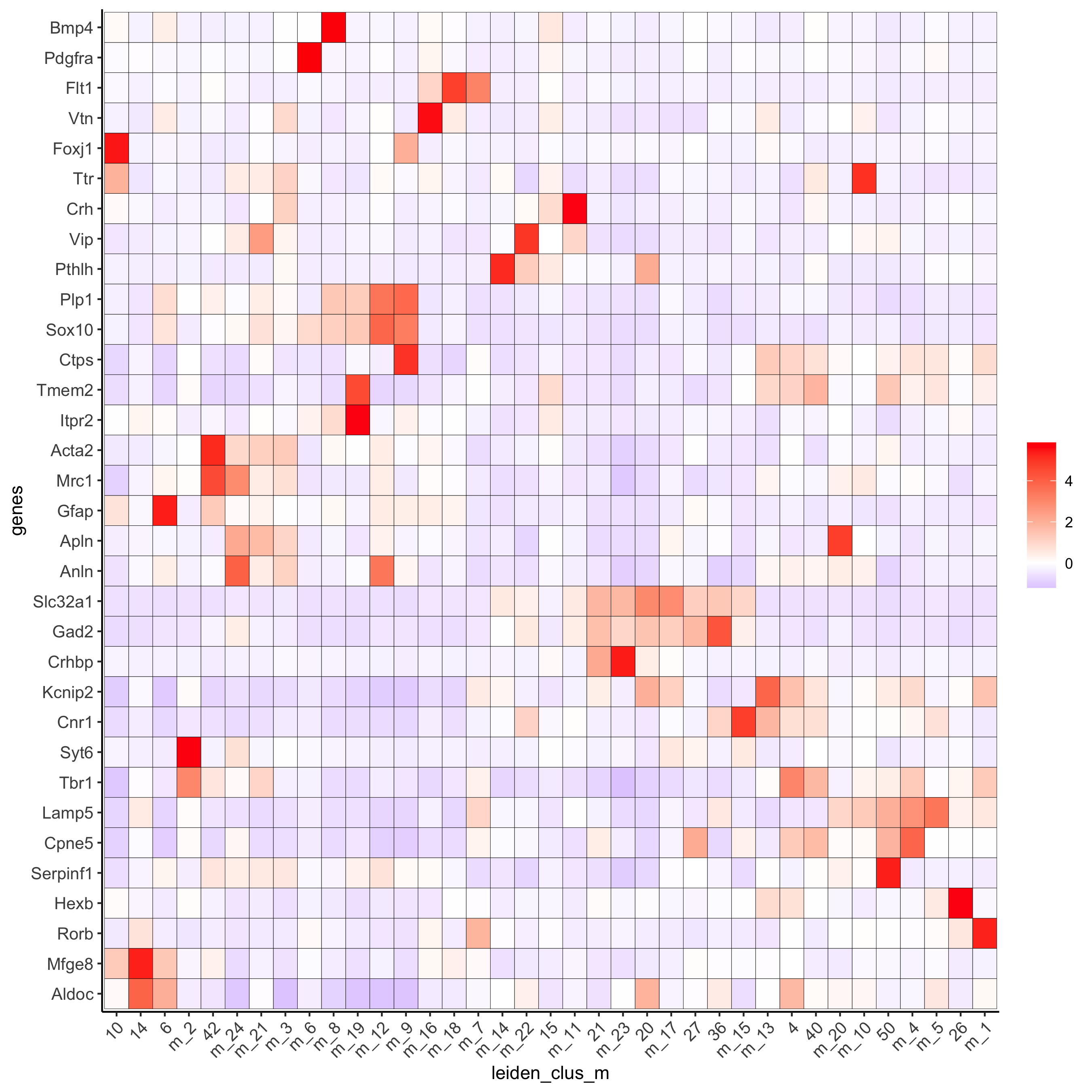
plotMetaDataHeatmap(osm_test, expression_values = 'custom', metadata_cols = c('leiden_clus_m'), save_param = c(save_name = '6_e_metaheatmap_all_genes'))
plotMetaDataHeatmap(osm_test, expression_values = 'custom', metadata_cols = c('ClusterName'), save_param = c(save_name = '6_f_metaheatmap_all_genes_names'))
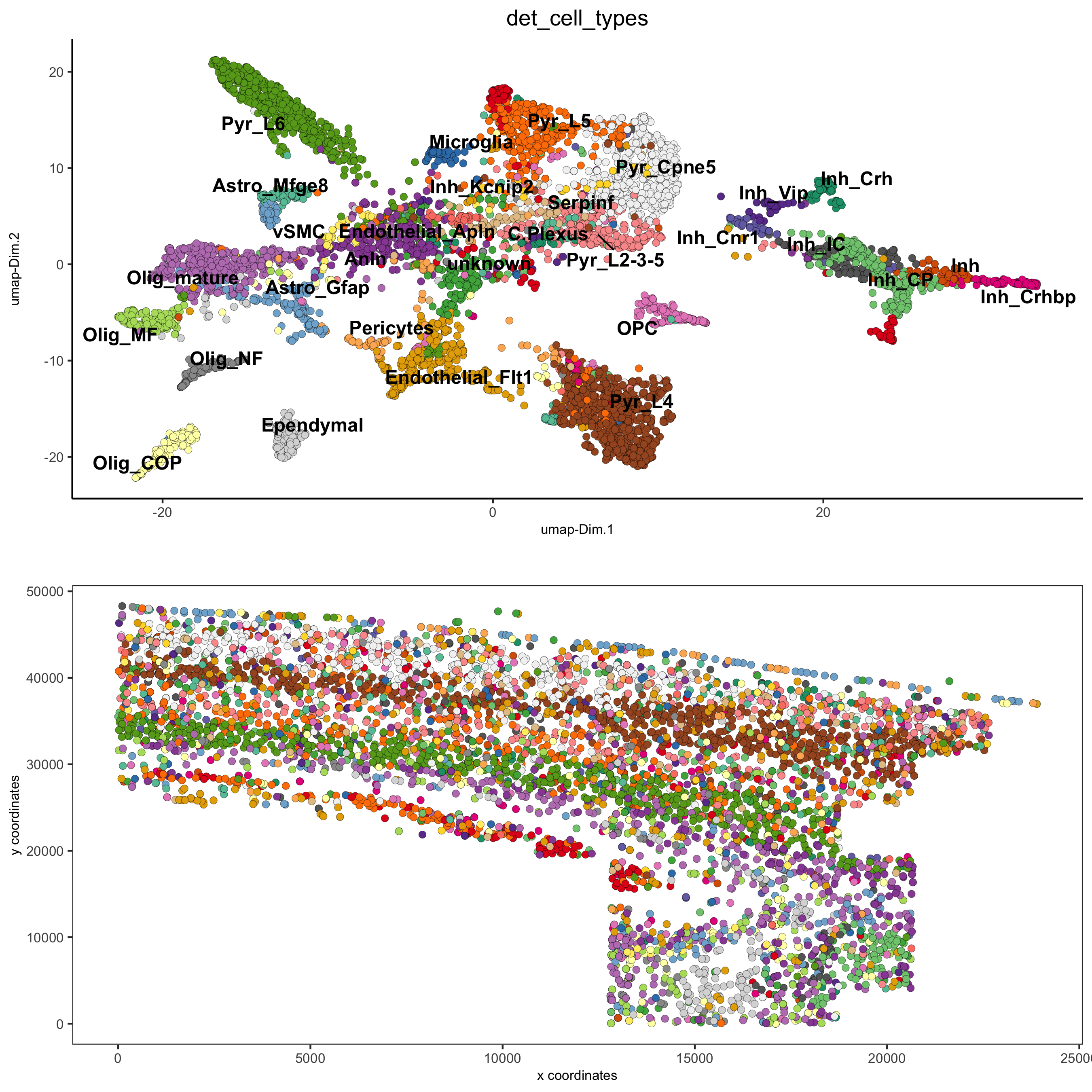
Part 7: Cell type annotation
## create vector with names ## compare clusters with osmFISH paper clusters_det_SS_cortex = c('Ependymal', 'Astro_Mfge8', 'Astro_Gfap', 'Pyr_L6', 'vSMC', 'Anln', 'Anln', 'Anln', 'OPC', 'Olig_COP', 'Olig_NF', 'Olig_mature', 'Olig_MF', 'Pericytes', 'Endothelial_Flt1', 'Endothelial_Flt1', 'Inh_Kcnip2', 'Inh_Vip', 'unknown', 'Inh_Crh', 'Inh', 'Inh_Crhbp', 'Inh_CP','Inh_CP', 'Inh_IC', 'Inh_IC', 'Inh_Cnr1', 'Inh_Kcnip2', 'Pyr_L5', 'Pyr_L5', 'Endothelial_Apln', 'C.Plexus', 'Serpinf', 'Pyr_Cpne5', 'Pyr_L2-3-5', 'Microglia', 'Pyr_L4') names(clusters_det_SS_cortex) = c('10', '14', '6', 'm_2', '42', 'm_24', 'm_21', 'm_3', 'm_6', 'm_8', 'm_19', 'm_12', 'm_9', 'm_16', 'm_18', 'm_7', 'm_14', 'm_22', '15', 'm_11', '21', 'm_23', '20', 'm_17', '27', '36', 'm_15', 'm_13', '4', '40', 'm_20', 'm_10', '50', 'm_4', 'm_5', '26', 'm_1') osm_test = annotateGiotto(gobject = osm_test, annotation_vector = clusters_det_SS_cortex, cluster_column = 'leiden_clus_m', name = 'det_cell_types') spatDimPlot2D(gobject = osm_test, cell_color = 'det_cell_types',dim_point_size = 2, spat_point_size = 2, save_param = c(save_name = '7_a_annotation_leiden_merged_detailed'))
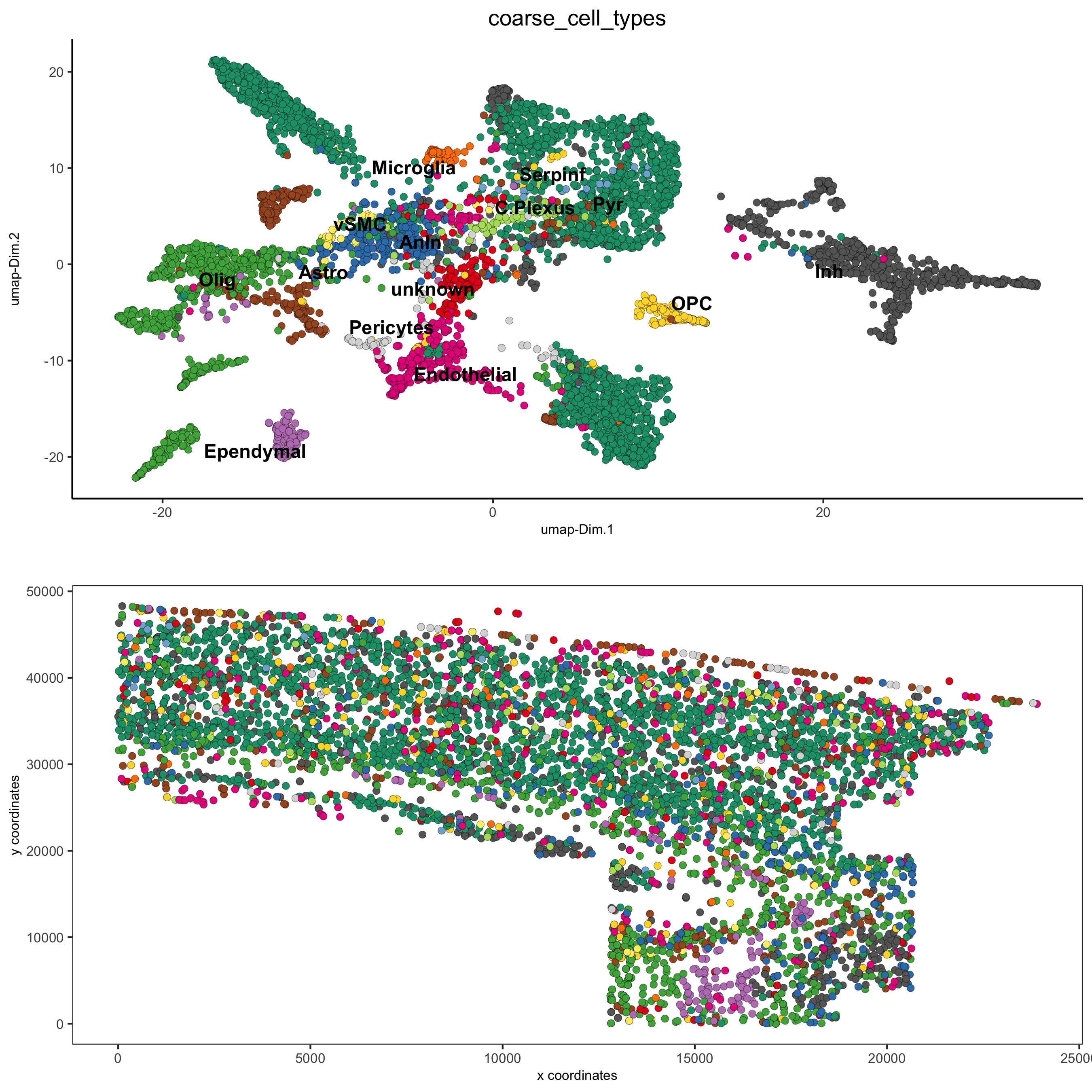
## coarse cell types clusters_coarse_SS_cortex = c('Ependymal', 'Astro', 'Astro', 'Pyr', 'vSMC', 'Anln', 'Anln', 'Anln', 'OPC', 'Olig', 'Olig', 'Olig', 'Olig', 'Pericytes', 'Endothelial', 'Endothelial', 'Inh', 'Inh', 'unknown', 'Inh', 'Inh', 'Inh', 'Inh', 'Inh', 'Inh', 'Inh', 'Inh', 'Inh', 'Pyr', 'Pyr', 'Endothelial', 'C.Plexus', 'Serpinf', 'Pyr', 'Pyr', 'Microglia', 'Pyr') names(clusters_coarse_SS_cortex) = c('Ependymal', 'Astro_Mfge8', 'Astro_Gfap', 'Pyr_L6', 'vSMC', 'Anln', 'Anln', 'Anln', 'OPC', 'Olig_COP', 'Olig_NF', 'Olig_mature', 'Olig_MF', 'Pericytes', 'Endothelial_Flt1', 'Endothelial_Flt1', 'Inh_Kcnip2', 'Inh_Vip', 'unknown', 'Inh_Crh', 'Inh', 'Inh_Crhbp', 'Inh_CP','Inh_CP', 'Inh_IC', 'Inh_IC', 'Inh_Cnr1', 'Inh_Kcnip2', 'Pyr_L5', 'Pyr_L5', 'Endothelial_Apln', 'C.Plexus', 'Serpinf', 'Pyr_Cpne5', 'Pyr_L2-3-5', 'Microglia', 'Pyr_L4') osm_test = annotateGiotto(gobject = osm_test, annotation_vector = clusters_coarse_SS_cortex, cluster_column = 'det_cell_types', name = 'coarse_cell_types') spatDimPlot2D(gobject = osm_test, cell_color = 'coarse_cell_types',dim_point_size = 1.5, spat_point_size = 1.5, save_param = c(save_name = '7_b_annotation_leiden_merged_coarse'))
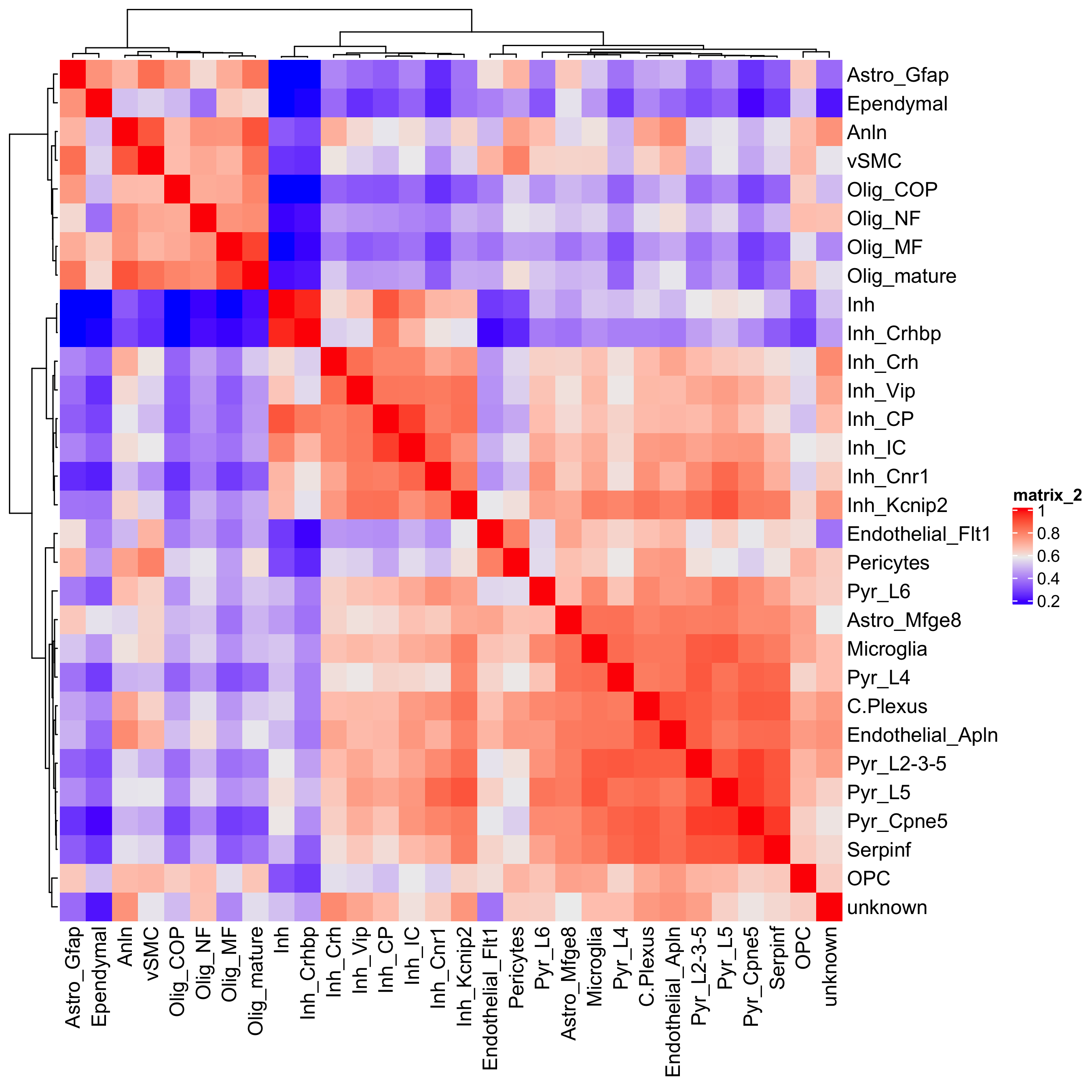
# heatmaps # showClusterHeatmap(gobject = osm_test, cluster_column = 'det_cell_types', save_param = c(save_name = '7_c_clusterHeatmap_det_cell_types', units = 'in'))
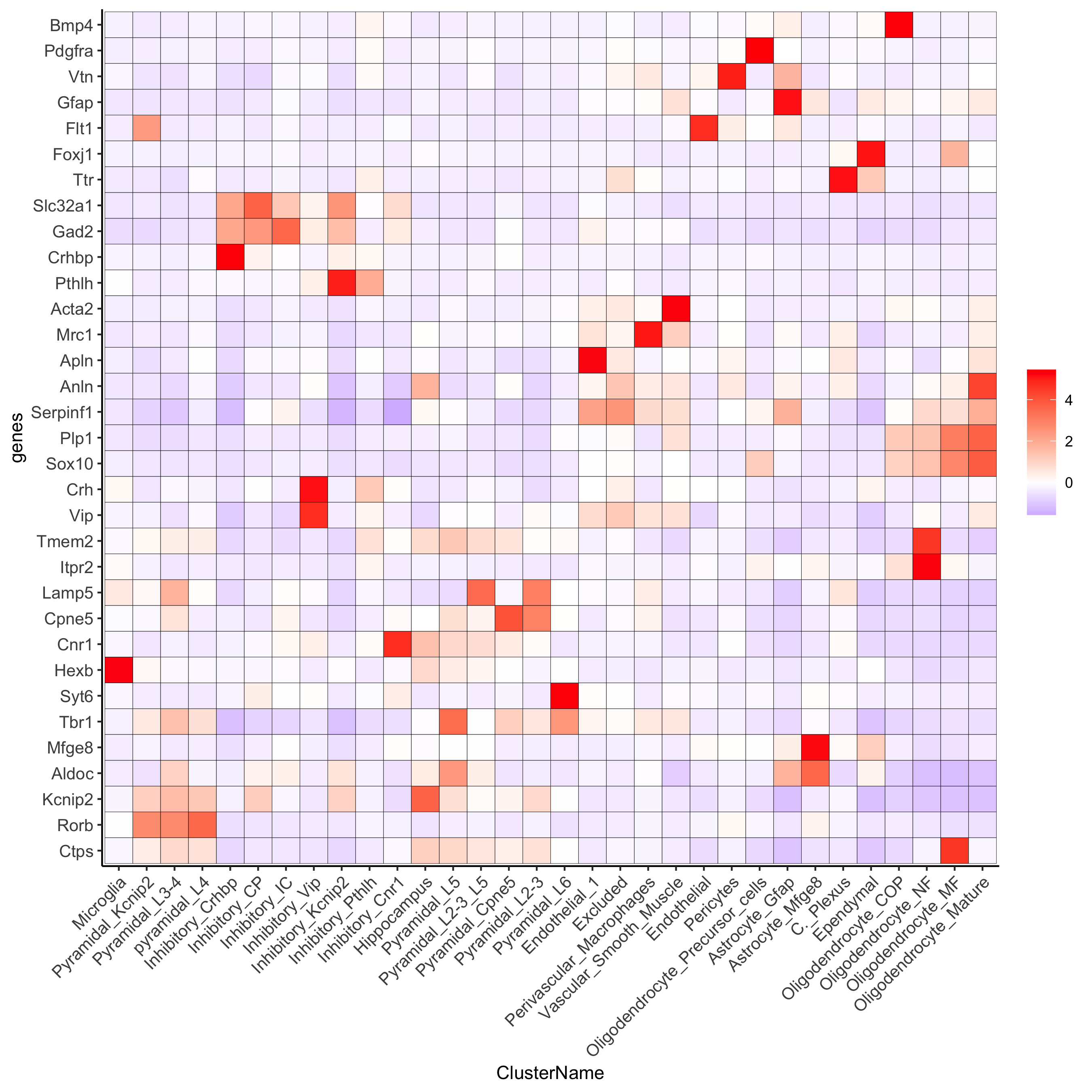
plotHeatmap(osm_test, genes = osm_test@gene_ID, cluster_column = 'det_cell_types', legend_nrows = 2, expression_values = 'custom', gene_order = 'correlation', cluster_order = 'correlation', save_param = c(save_name = '7_d_heatamp_det_cell_types'))
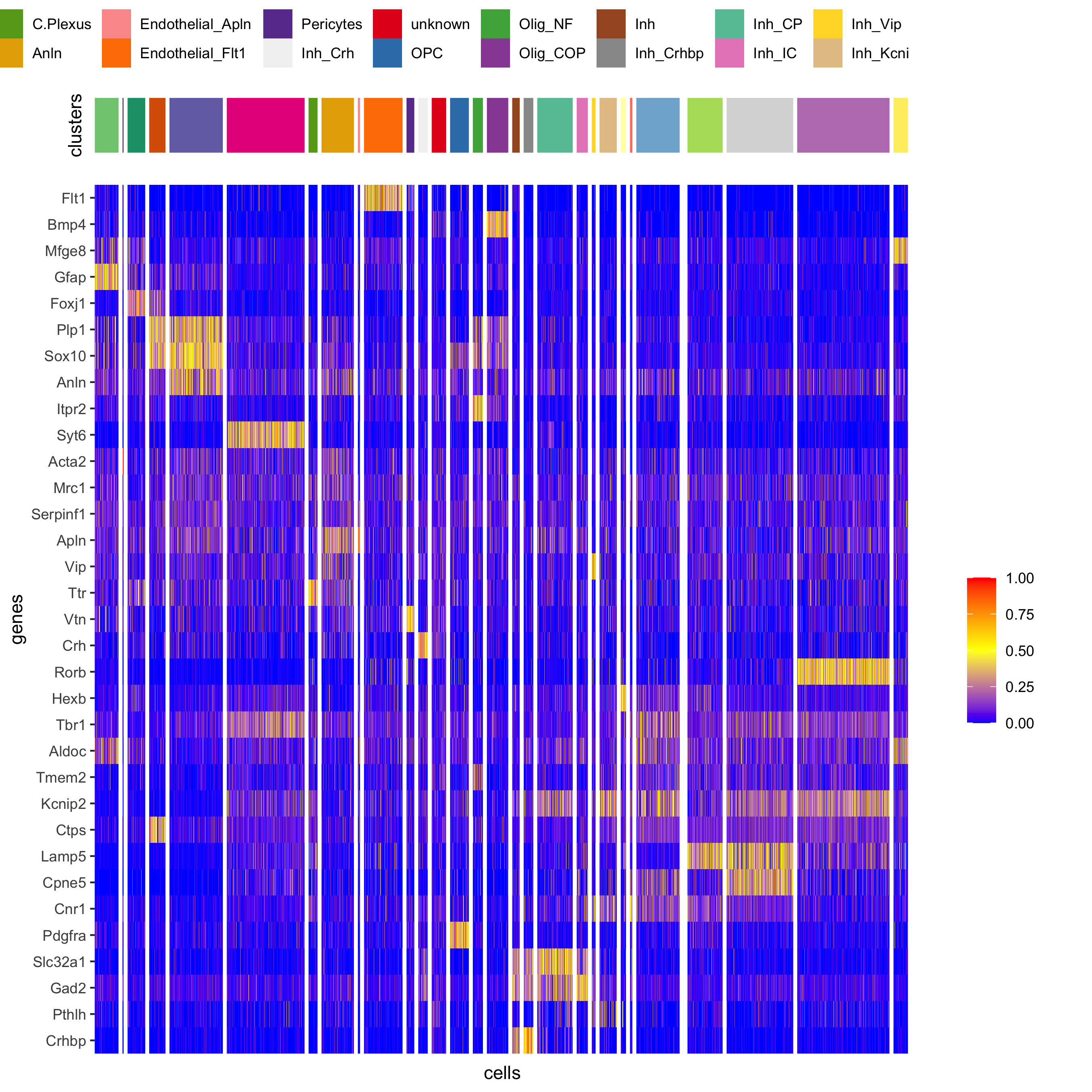
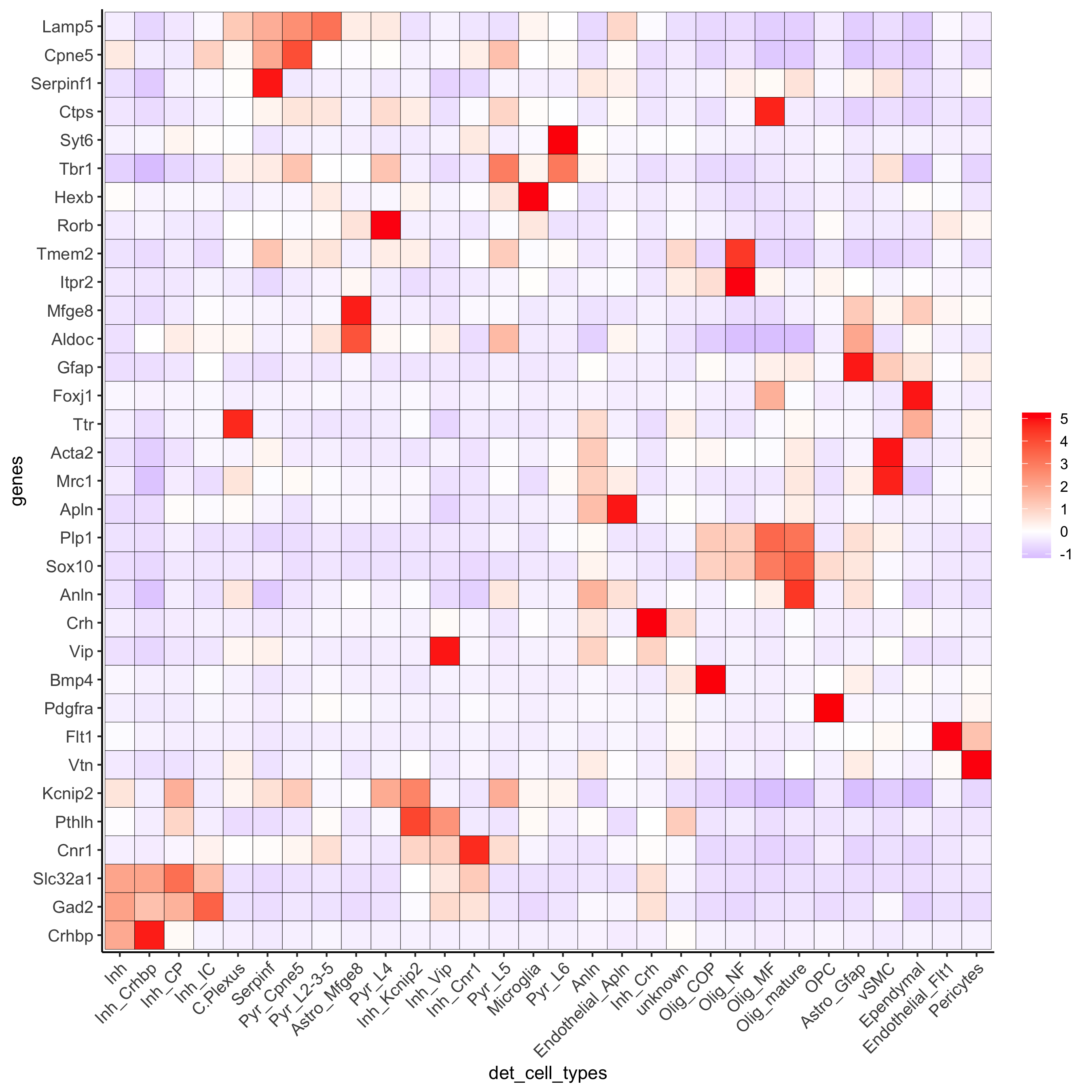
plotMetaDataHeatmap(osm_test, expression_values = 'custom', metadata_cols = c('det_cell_types'), save_param = c(save_name = '7_e_metaheatmap'))
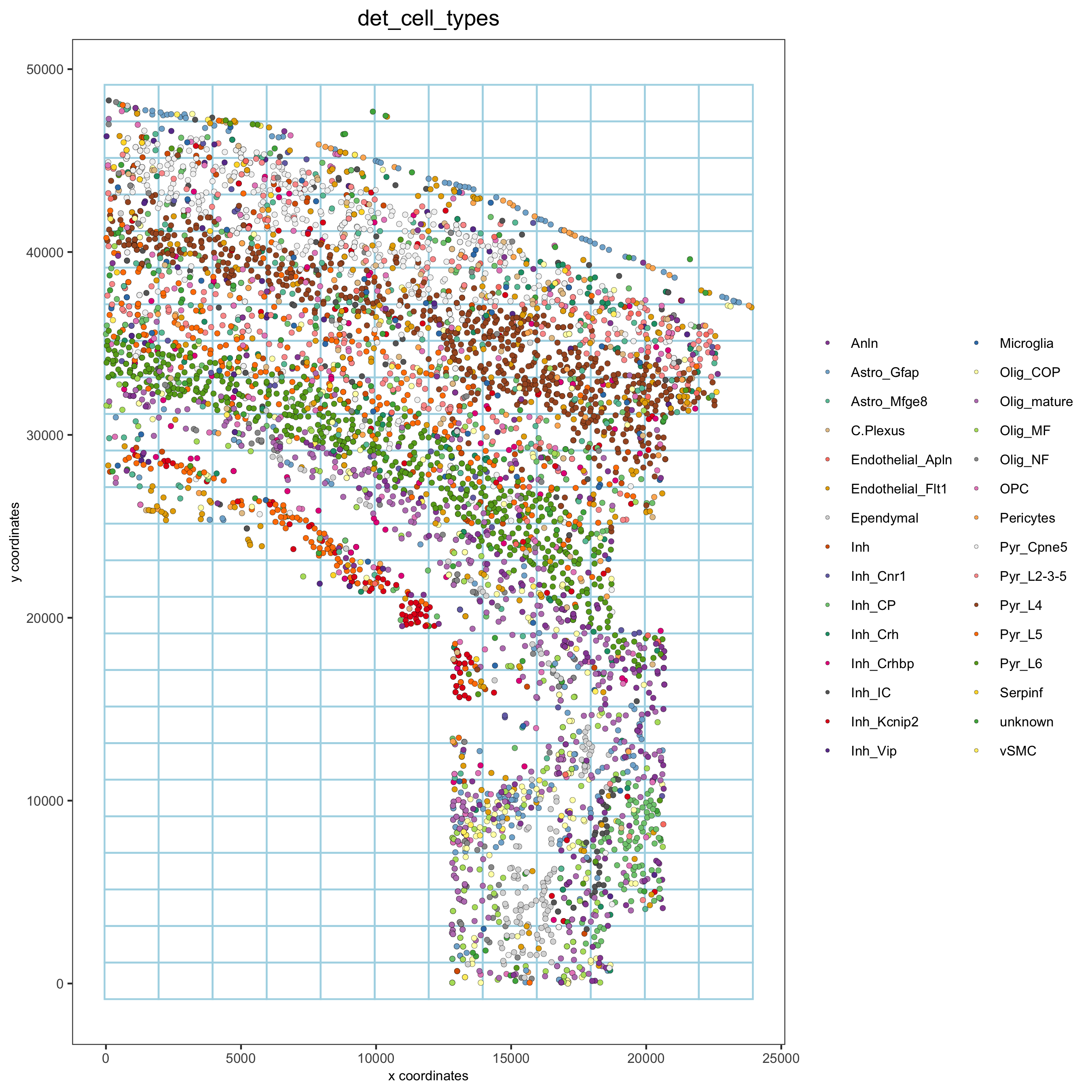
Part 8: Spatial grid
osm_test <- createSpatialGrid(gobject = osm_test, sdimx_stepsize = 2000, sdimy_stepsize = 2000, minimum_padding = 0) spatPlot2D(osm_test, cell_color = 'det_cell_types', show_grid = T, grid_color = 'lightblue', spatial_grid_name = 'spatial_grid', point_size = 1.5, save_param = c(save_name = '8_grid_det_cell_types'))
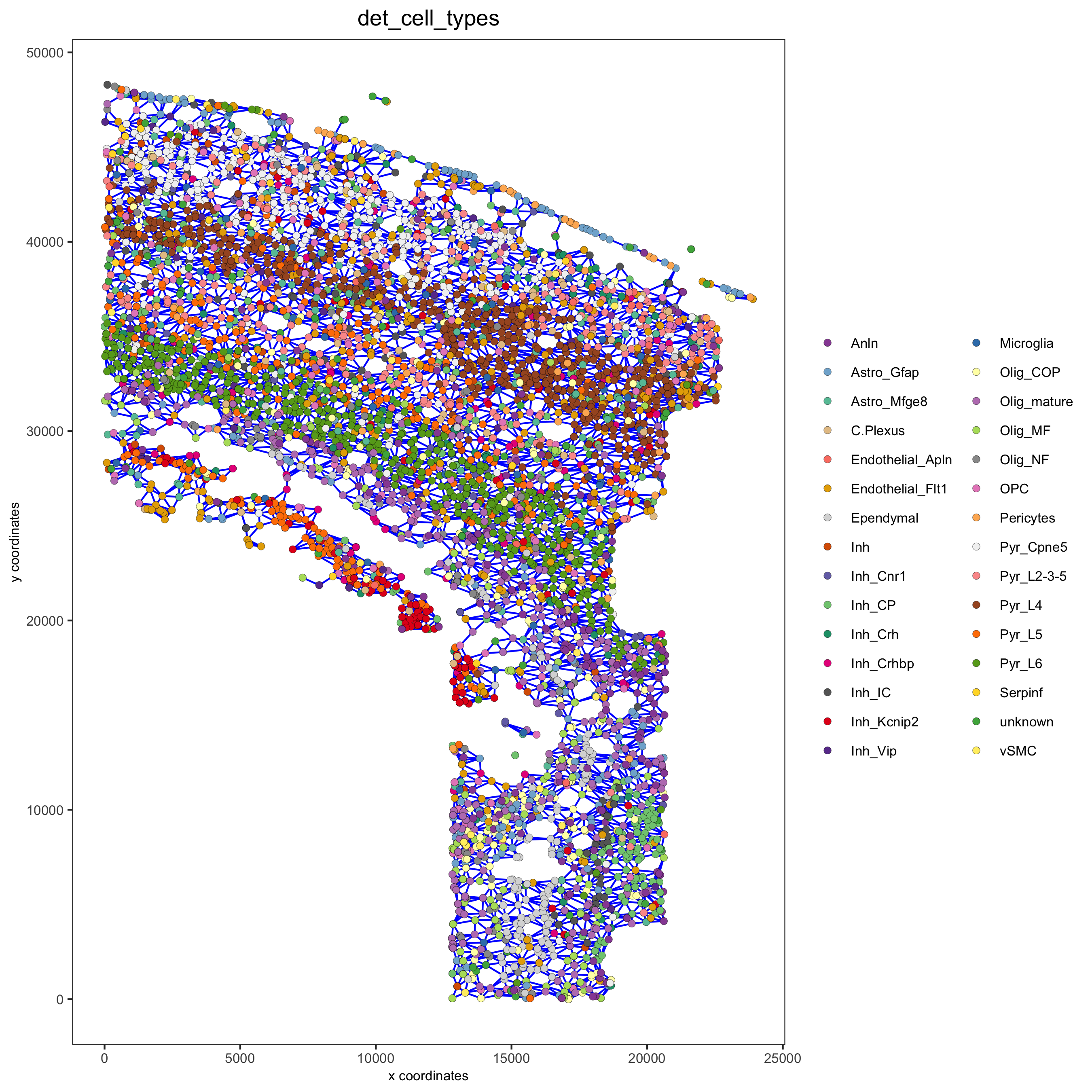
Part 9: Spatial network
osm_test <- createSpatialNetwork(gobject = osm_test) spatPlot2D(gobject = osm_test, show_network = T, network_color = 'blue', point_size = 1.5, cell_color = 'det_cell_types', legend_symbol_size = 2, save_param = c(save_name = '9_spatial_network_k10'))
Part 10: Spatial genes
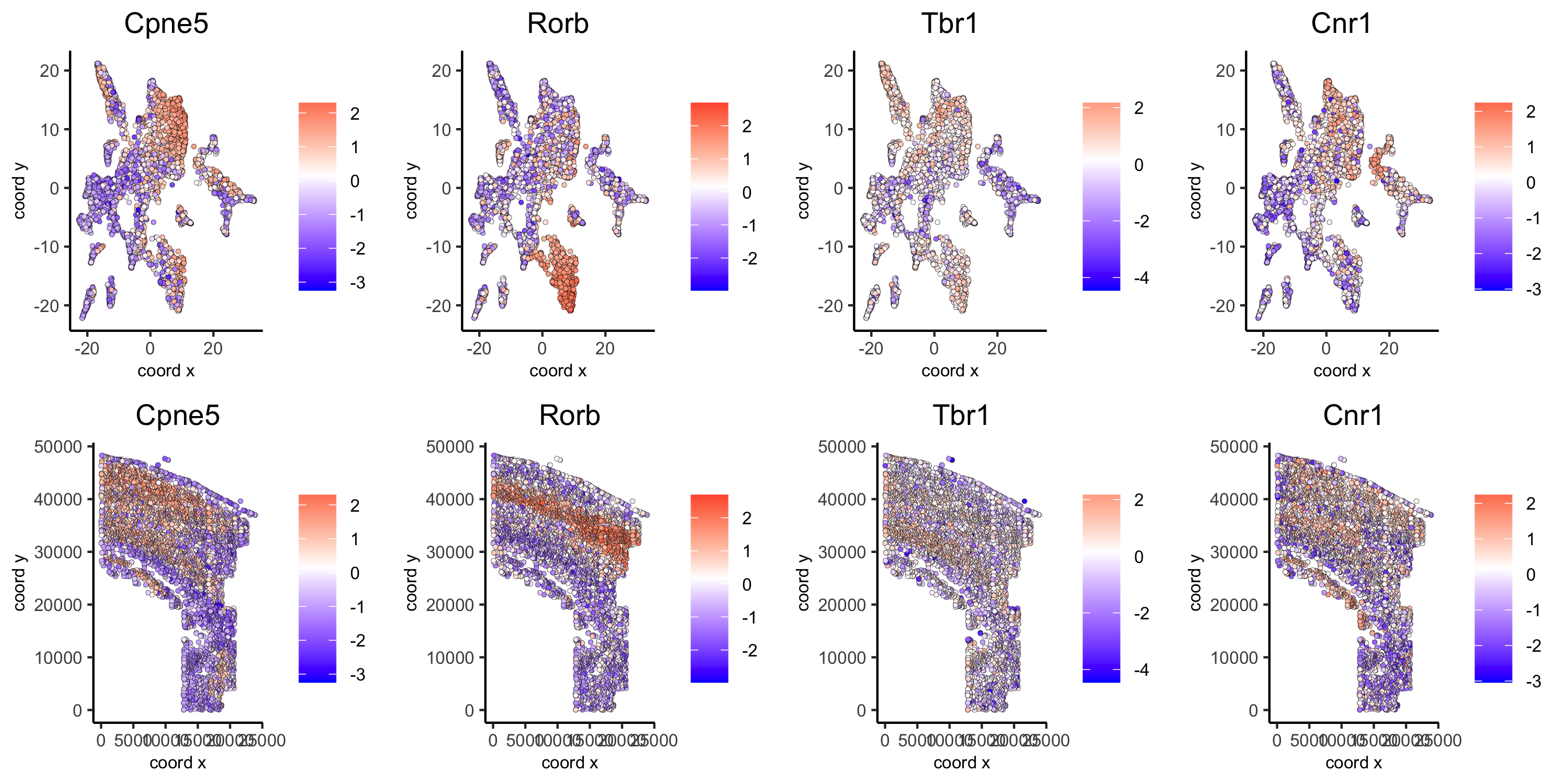
# km binarization kmtest = binSpect(osm_test, bin_method = 'kmeans') spatDimGenePlot2D(osm_test, expression_values = 'scaled', genes = kmtest$genes[1:4], plot_alignment = 'vertical', cow_n_col = 4, save_param = c(save_name = '10_a_spatial_genes_km', base_height = 5, base_width = 10))
Part 12. cell-cell preferential proximity
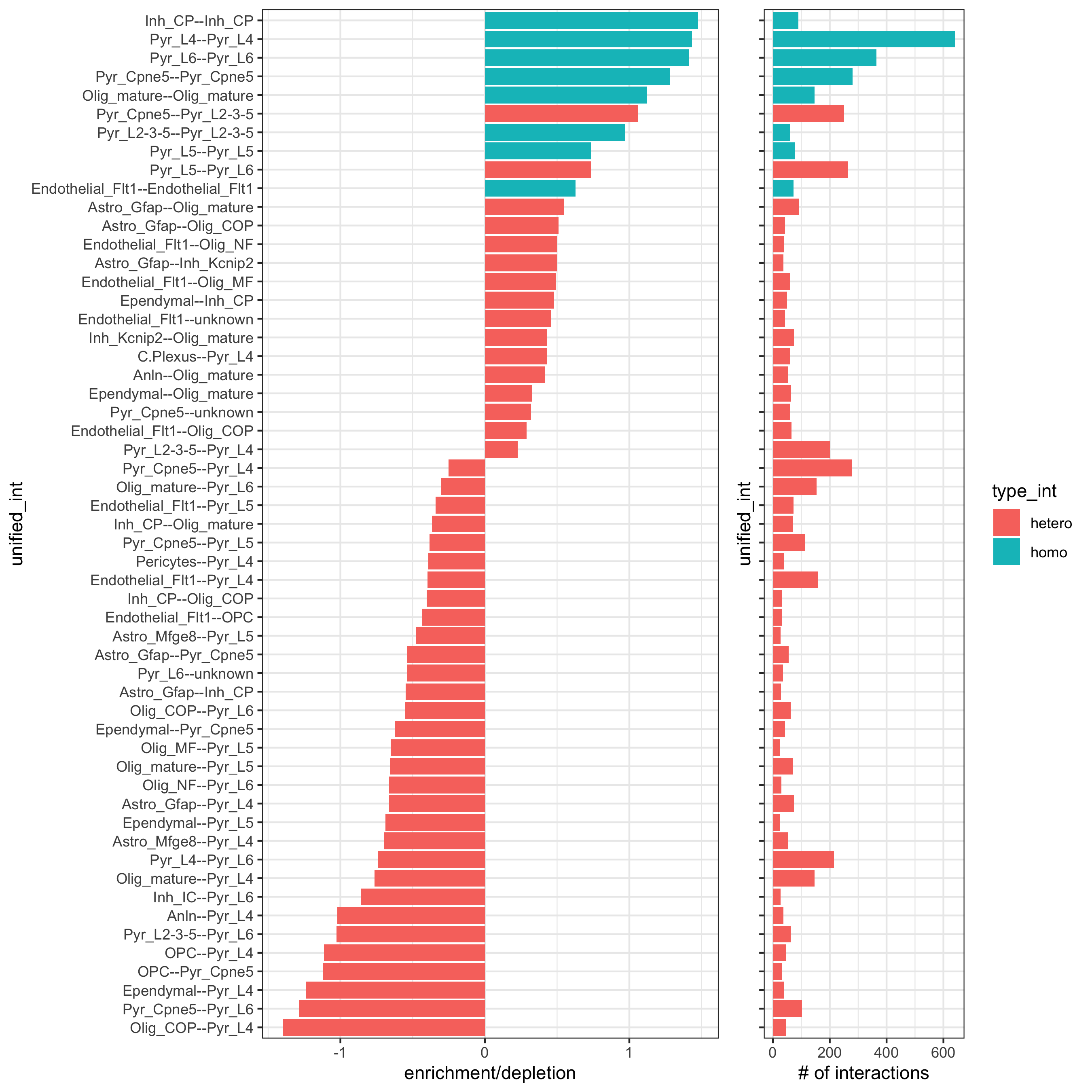
## calculate frequently seen proximities cell_proximities = cellProximityEnrichment(gobject = osm_test, cluster_column = 'det_cell_types', number_of_simulations = 1000) ## barplot cellProximityBarplot(gobject = osm_test, CPscore = cell_proximities, min_orig_ints = 25, min_sim_ints = 25, save_param = c(save_name = '12_a_barplot_cell_cell_enrichment'))
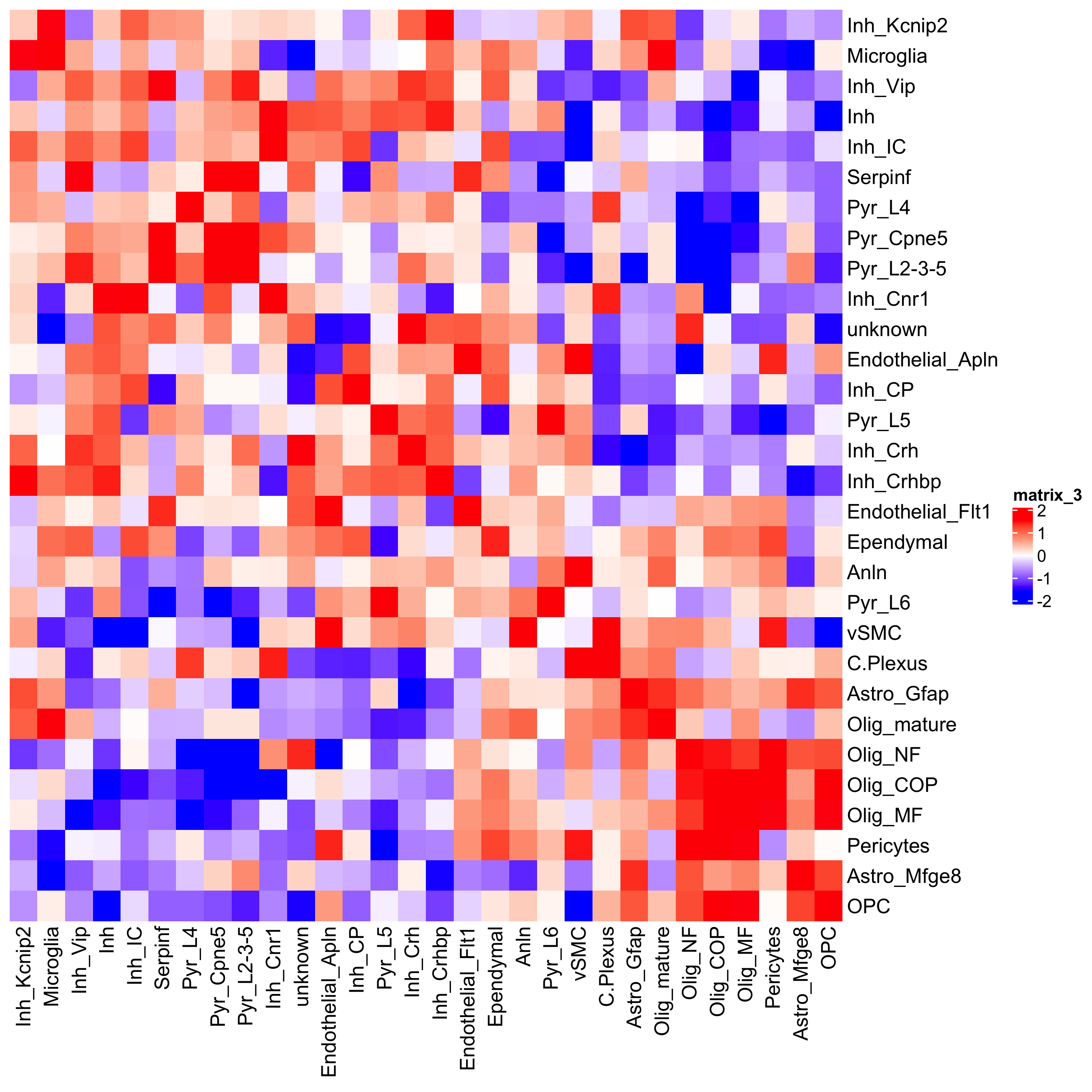
## heatmap cellProximityHeatmap(gobject = osm_test, CPscore = cell_proximities, order_cell_types = T, scale = T, color_breaks = c(-1.5, 0, 1.5), color_names = c('blue', 'white', 'red'), save_param = c(save_name = '12_b_heatmap_cell_cell_enrichment', unit = 'in'))
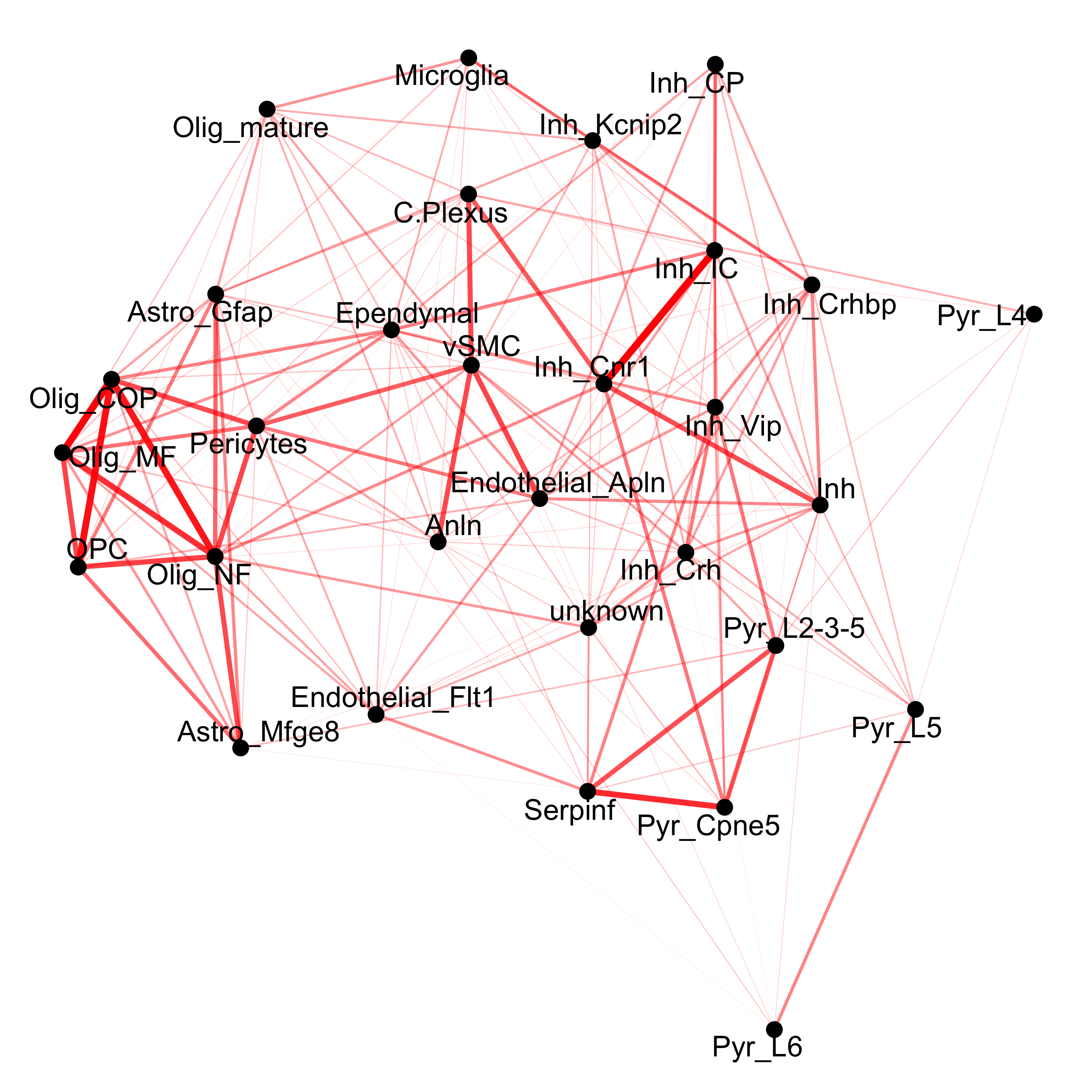
## network cellProximityNetwork(gobject = osm_test, CPscore = cell_proximities, remove_self_edges = T, only_show_enrichment_edges = T, save_param = c(save_name = '12_c_network_cell_cell_enrichment'))
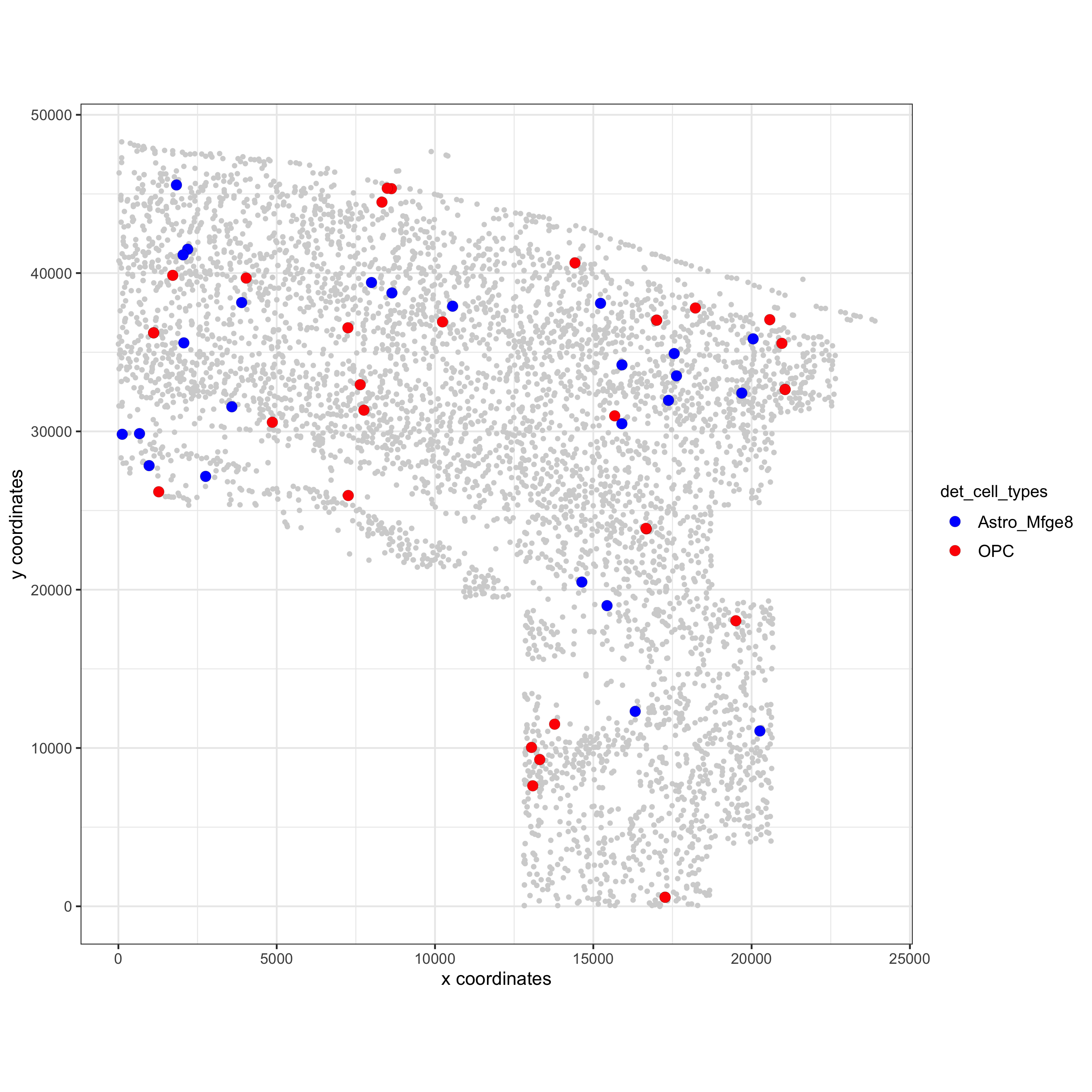
## visualization spec_interaction = "Astro_Mfge8--OPC" cellProximitySpatPlot(gobject = osm_test, interaction_name = spec_interaction, cluster_column = 'det_cell_types', cell_color = 'det_cell_types', cell_color_code = c('Astro_Mfge8' = 'blue', 'OPC' = 'red'), coord_fix_ratio = 0.5, point_size_select = 3, point_size_other = 1.5, save_param = c(save_name = '12_d_cell_cell_enrichment_selected'))