Install Python modules
To run this vignette you need to install all the necessary Python modules.
This can be done manually, see https://rubd.github.io/Giotto_site/articles/installation_issues.html#python-manual-installation
This can be done within R using our installation tools (installGiottoEnvironment), see https://rubd.github.io/Giotto_site/articles/tut0_giotto_environment.html for more information.
(optional) set giotto instructions
# to automatically save figures in save_dir set save_plot to TRUE temp_dir = '~/Temp/' myinstructions = createGiottoInstructions(save_dir = temp_dir, save_plot = TRUE, show_plot = FALSE) # set python_path if you want to or install giotto environment python_path = NULL if(is.null(python_path)) { installGiottoEnvironment() }
1. Create a Giotto object
minimum requirements:
- matrix with expression information (or path to)
- x,y(,z) coordinates for cells or spots (or path to)
# giotto object expr_path = system.file("extdata", "starmap_expr.txt", package = 'Giotto') loc_path = system.file("extdata", "starmap_cell_loc.txt", package = 'Giotto') starmap_mini <- createGiottoObject(raw_exprs = expr_path, spatial_locs = loc_path, instructions = myinstructions)
How to work with Giotto instructions that are part of your Giotto object:
- show the instructions associated with your Giotto object with showGiottoInstructions
- change one or more instructions with changeGiottoInstructions
- replace all instructions at once with replaceGiottoInstructions
- read or get a specific giotto instruction with readGiottoInstructions
Of note, the python path can only be set once in an R session. See the reticulate package for more information.
# show instructions associated with giotto object (starmap_mini) showGiottoInstructions(starmap_mini)
2. processing steps
- filter genes and cells based on detection frequencies
- normalize expression matrix (log transformation, scaling factor and/or z-scores)
- add cell and gene statistics (optional)
- adjust expression matrix for technical covariates or batches (optional). These results will be stored in the custom slot.
filterDistributions(starmap_mini, detection = 'genes', save_param = list(save_name = '2_a_filtergenes'))
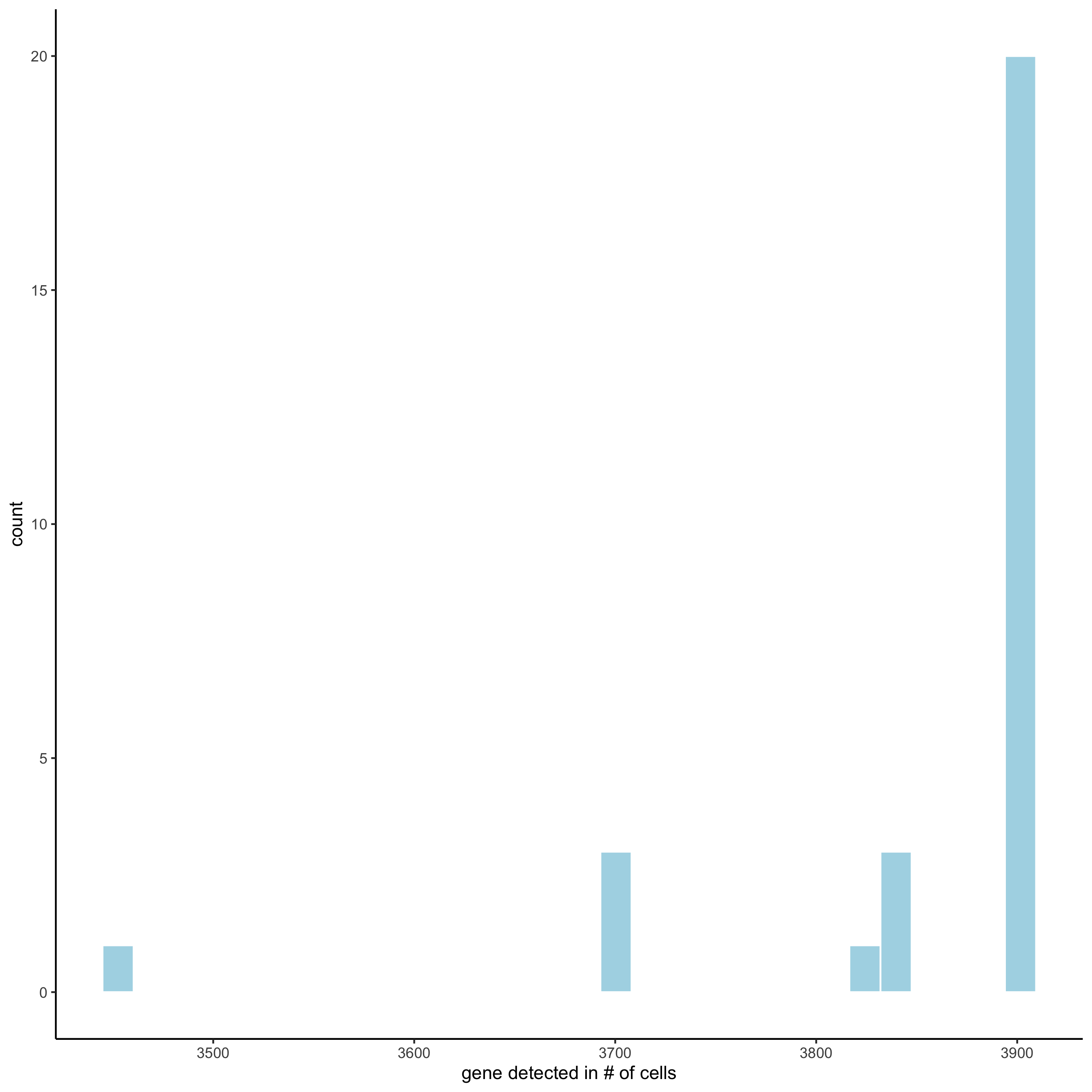
filterDistributions(starmap_mini, detection = 'cells', save_param = list(save_name = '2_b_filtercells'))
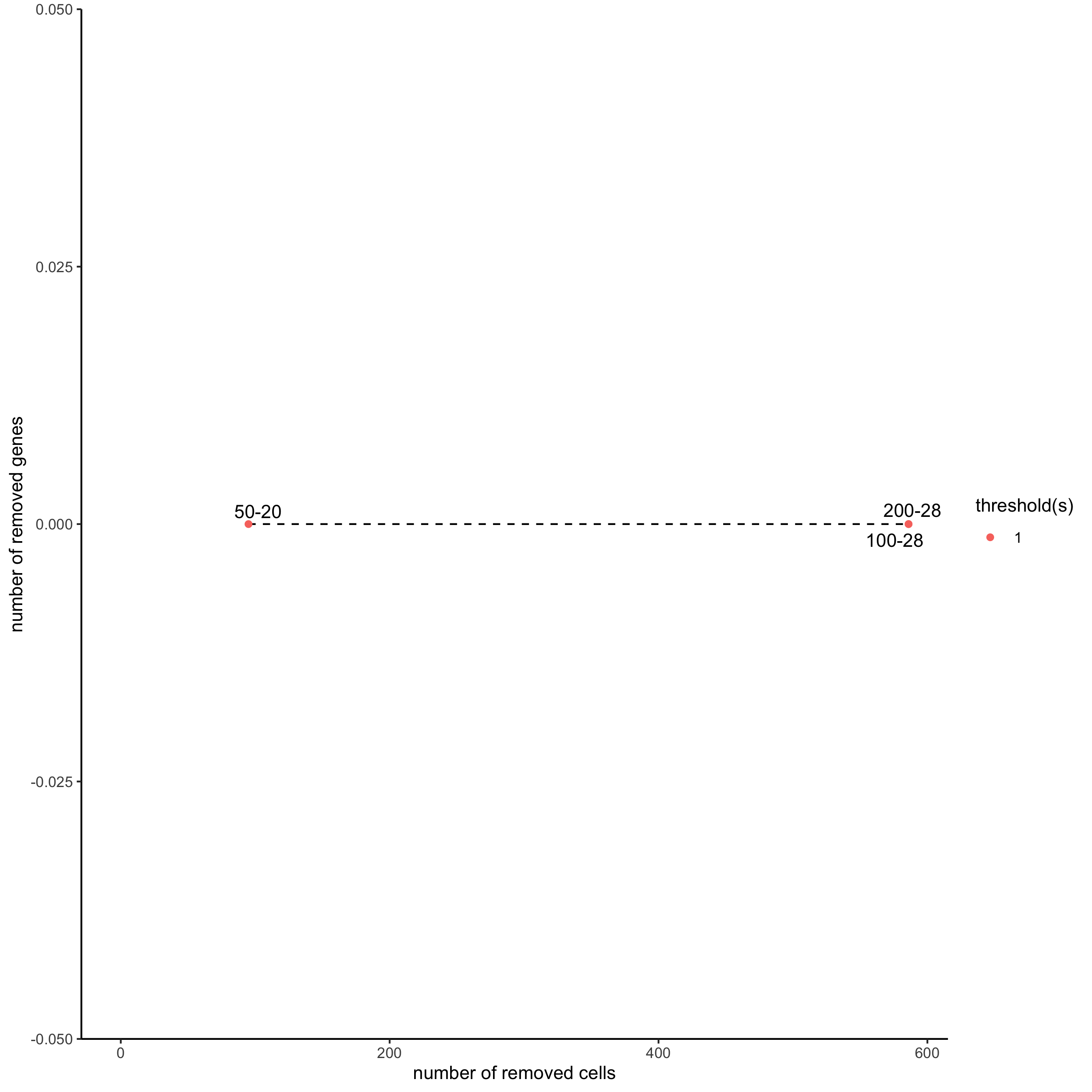
filterCombinations(starmap_mini, expression_thresholds = c(1), gene_det_in_min_cells = c(50, 100, 200), min_det_genes_per_cell = c(20, 28, 28), save_param = list(save_name = '2_c_filtercombos'))
starmap_mini <- filterGiotto(gobject = starmap_mini, expression_threshold = 1, gene_det_in_min_cells = 50, min_det_genes_per_cell = 20, expression_values = c('raw'), verbose = T) starmap_mini <- normalizeGiotto(gobject = starmap_mini, scalefactor = 6000, verbose = T) starmap_mini <- addStatistics(gobject = starmap_mini)
3. dimension reduction
- identify highly variable genes (HVG) will not be performed here, because there are only few genes
- perform PCA
- identify number of significant prinicipal components (PCs)
- run UMAP and/or TSNE on PCs (or directly on matrix)
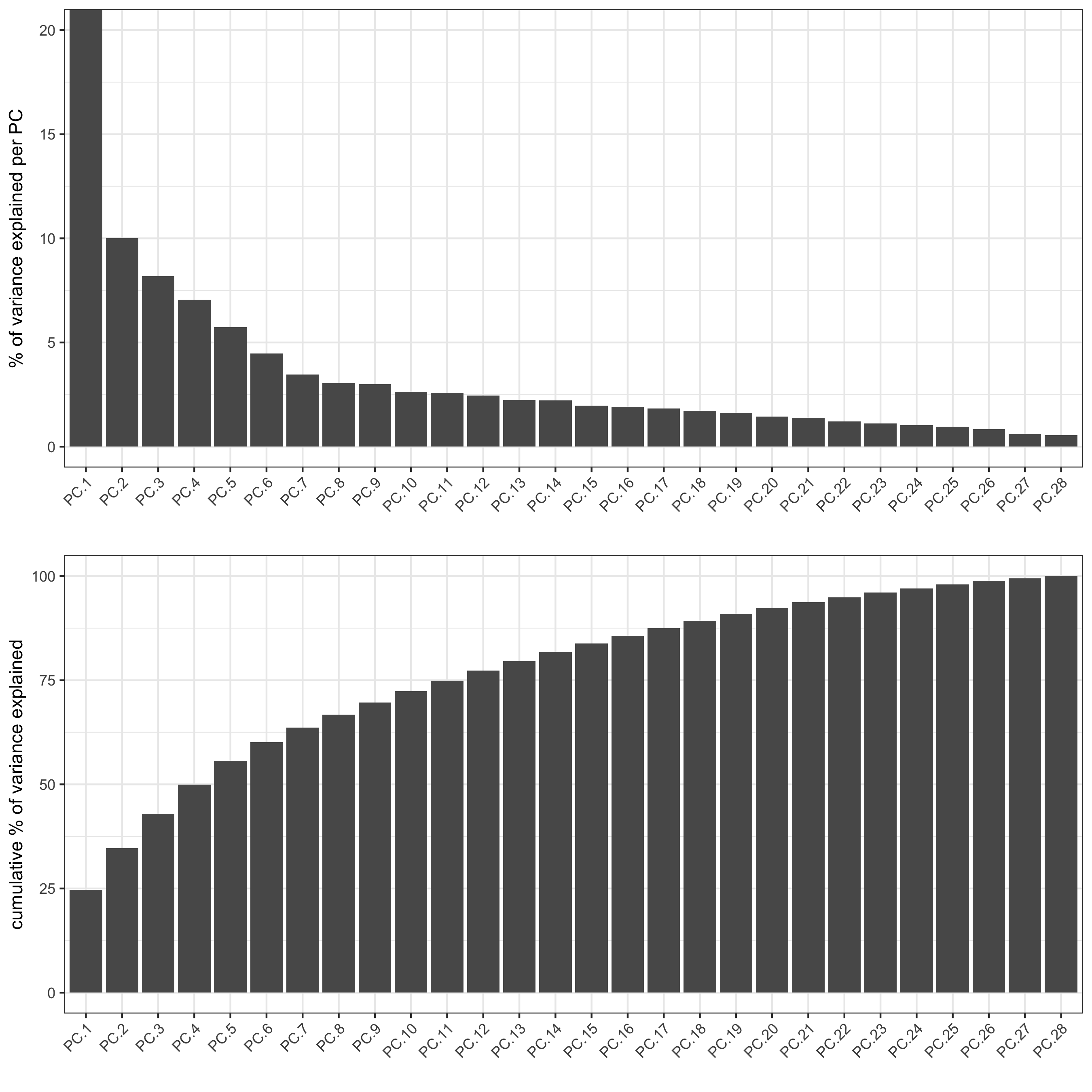
starmap_mini <- runPCA(gobject = starmap_mini, method = 'factominer') screePlot(starmap_mini, ncp = 30, save_param = list(save_name = '3_a_screeplot'))
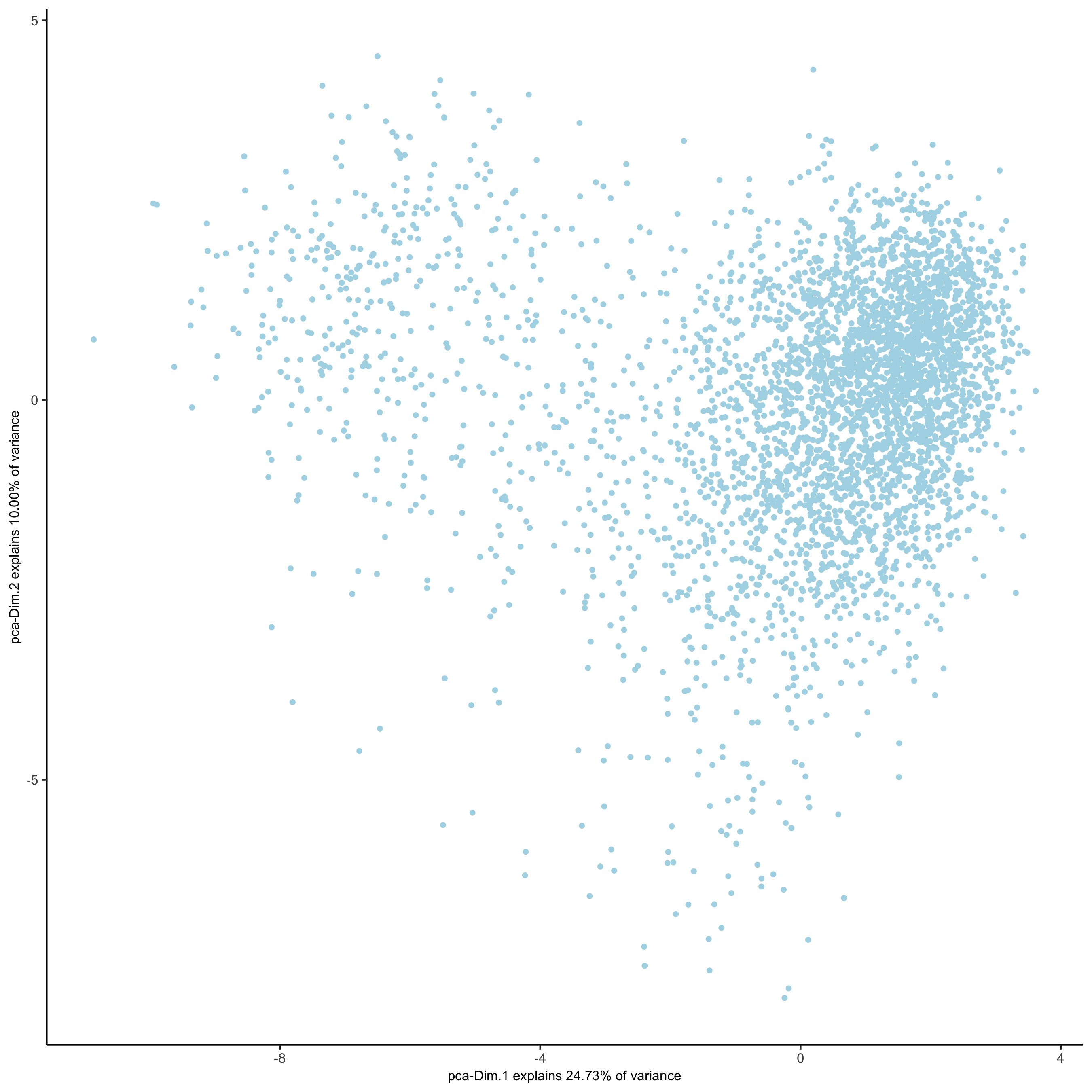
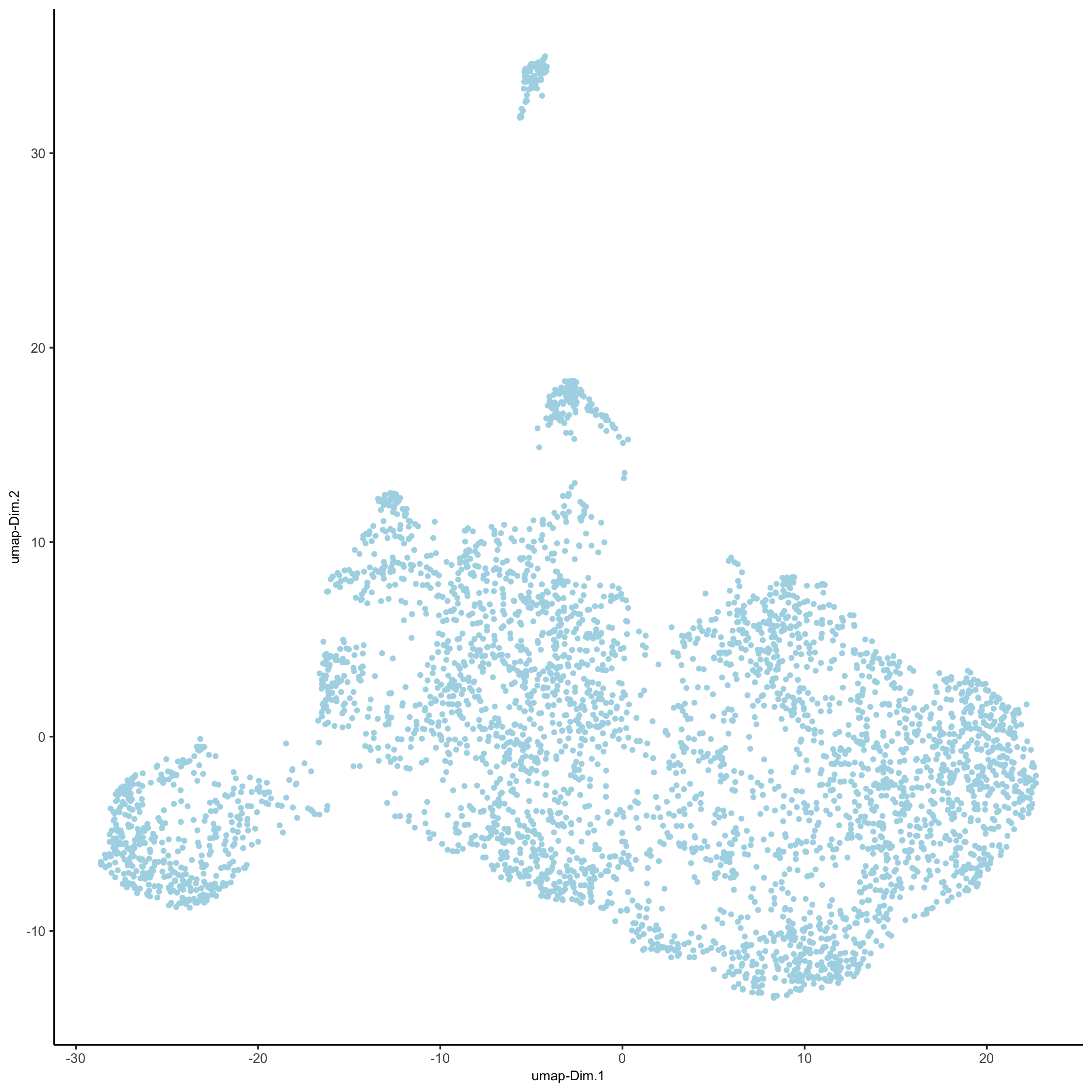
# 2D umap starmap_mini <- runUMAP(starmap_mini, dimensions_to_use = 1:8) plotUMAP(gobject = starmap_mini, save_param = list(save_name = '3_c_UMAP'))
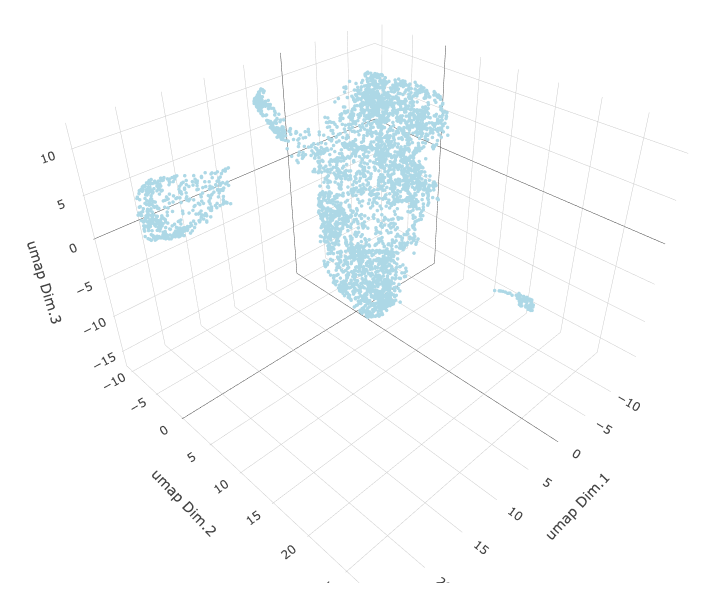
# 3D umap starmap_mini <- runUMAP(starmap_mini, dimensions_to_use = 1:8, name = '3D_umap', n_components = 3) plotUMAP_3D(gobject = starmap_mini, dim_reduction_name = '3D_umap', save_param = list(save_name = '3_d_UMAP_3D'))
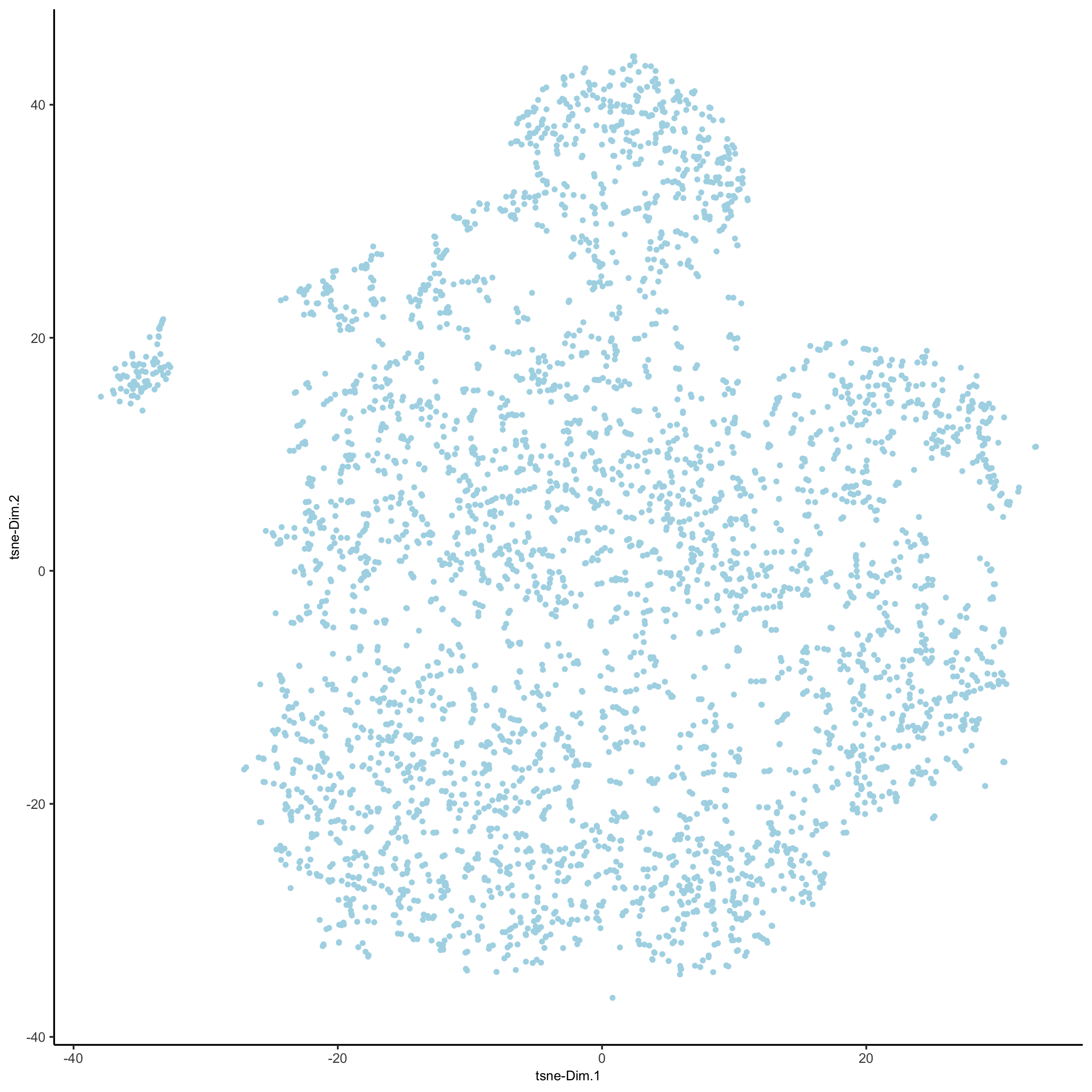
# 2D tsne starmap_mini <- runtSNE(starmap_mini, dimensions_to_use = 1:8) plotTSNE(gobject = starmap_mini, save_param = list(save_name = '3_e_TSNE'))
4. clustering
- create a shared (default) nearest network in PCA space (or directly on matrix)
- cluster on nearest network with Leiden or Louvan (kmeans and hclust are alternatives)
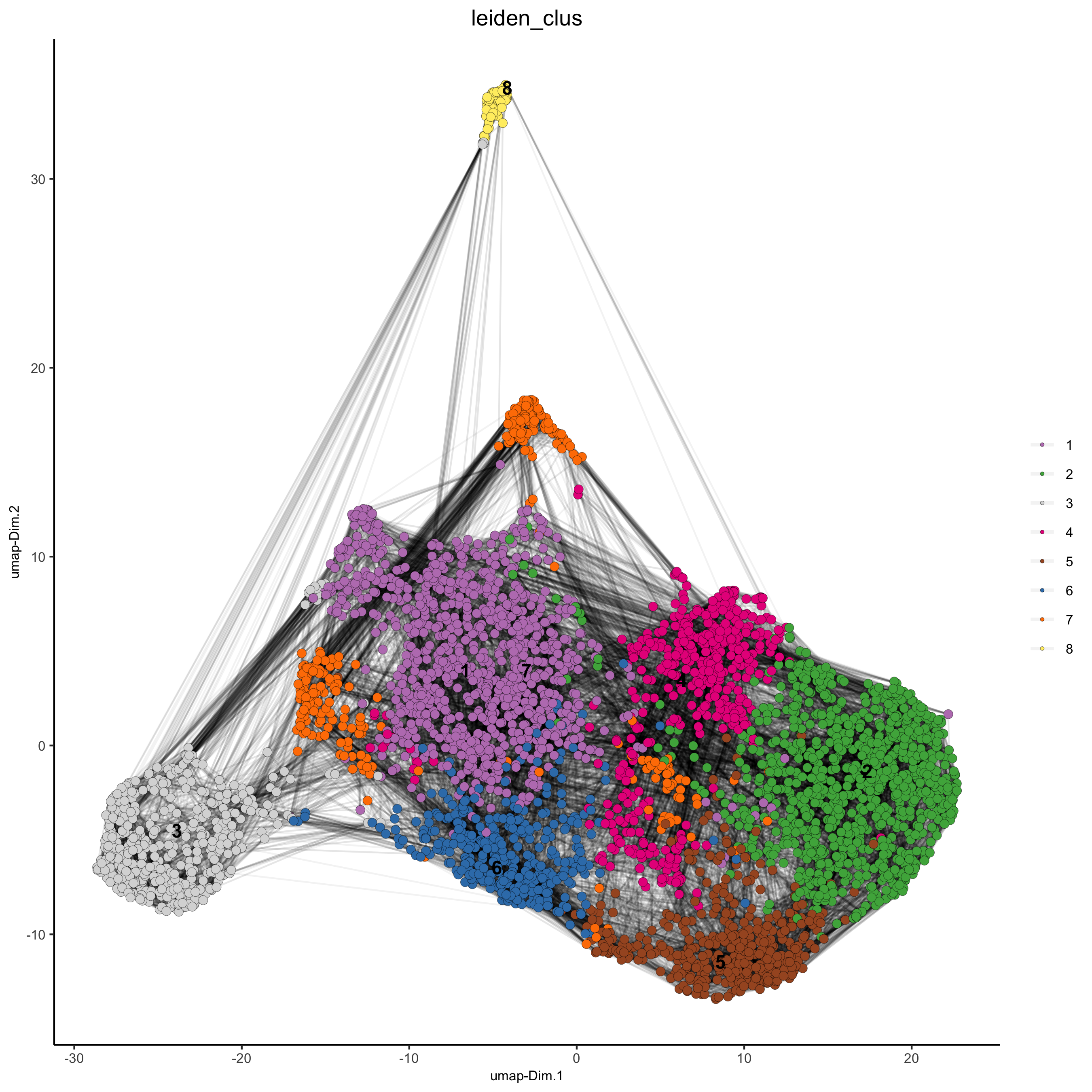
starmap_mini <- createNearestNetwork(gobject = starmap_mini, dimensions_to_use = 1:8, k = 25) starmap_mini <- doLeidenCluster(gobject = starmap_mini, resolution = 0.5, n_iterations = 1000) # 2D umap plotUMAP(gobject = starmap_mini, cell_color = 'leiden_clus', show_NN_network = T, point_size = 2.5, save_param = list(save_name = '4_a_UMAP'))
# 3D umap plotUMAP_3D(gobject = starmap_mini, dim_reduction_name = '3D_umap', cell_color = 'leiden_clus', save_param = list(save_name = '4_b_UMAP_3D'))
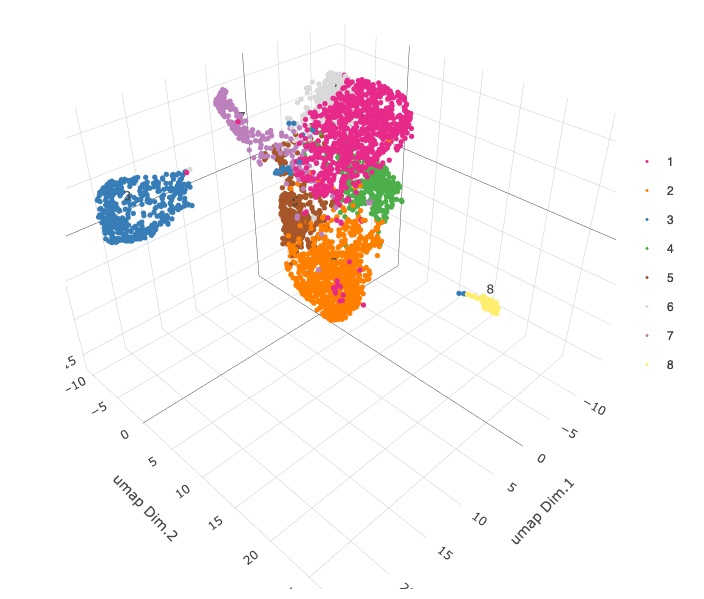
# 2D umap + coordinates spatDimPlot(gobject = starmap_mini, cell_color = 'leiden_clus', dim_point_size = 2, spat_point_size = 2.5, save_param = list(save_name = '4_c_spatdimplot'))

# 3D umap + coordinates spatDimPlot3D(gobject = starmap_mini, cell_color = 'leiden_clus', dim_reduction_name = '3D_umap', save_param = list(save_name = '4_d_spatdimplot3D'))
# heatmap and dendrogram showClusterHeatmap(gobject = starmap_mini, cluster_column = 'leiden_clus', save_param = list(save_name = '4_e_clusterheatmap'))

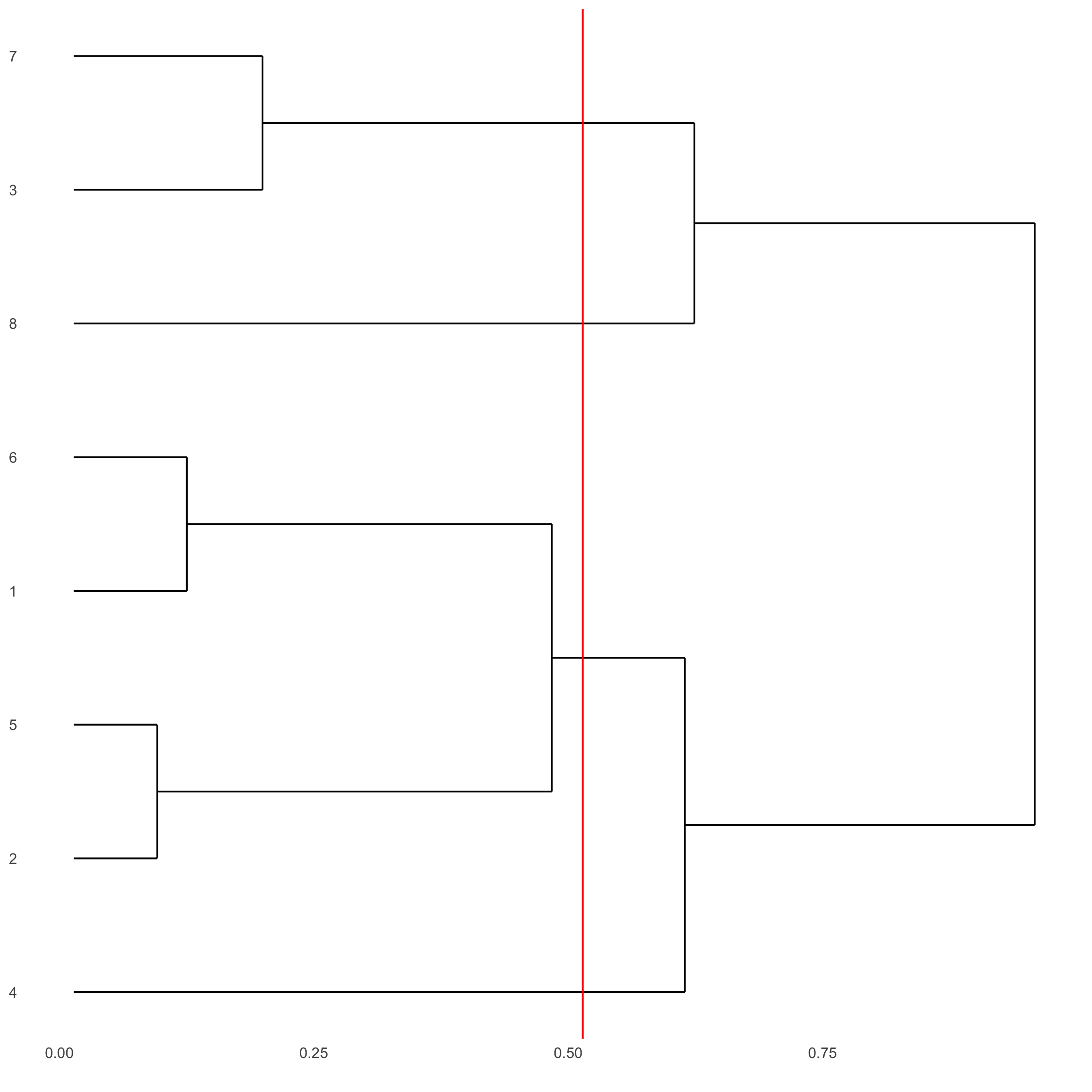
showClusterDendrogram(starmap_mini, h = 0.5, rotate = T, cluster_column = 'leiden_clus', save_param = list(save_name = '4_f_clusterdendrogram'))
5. differential expression
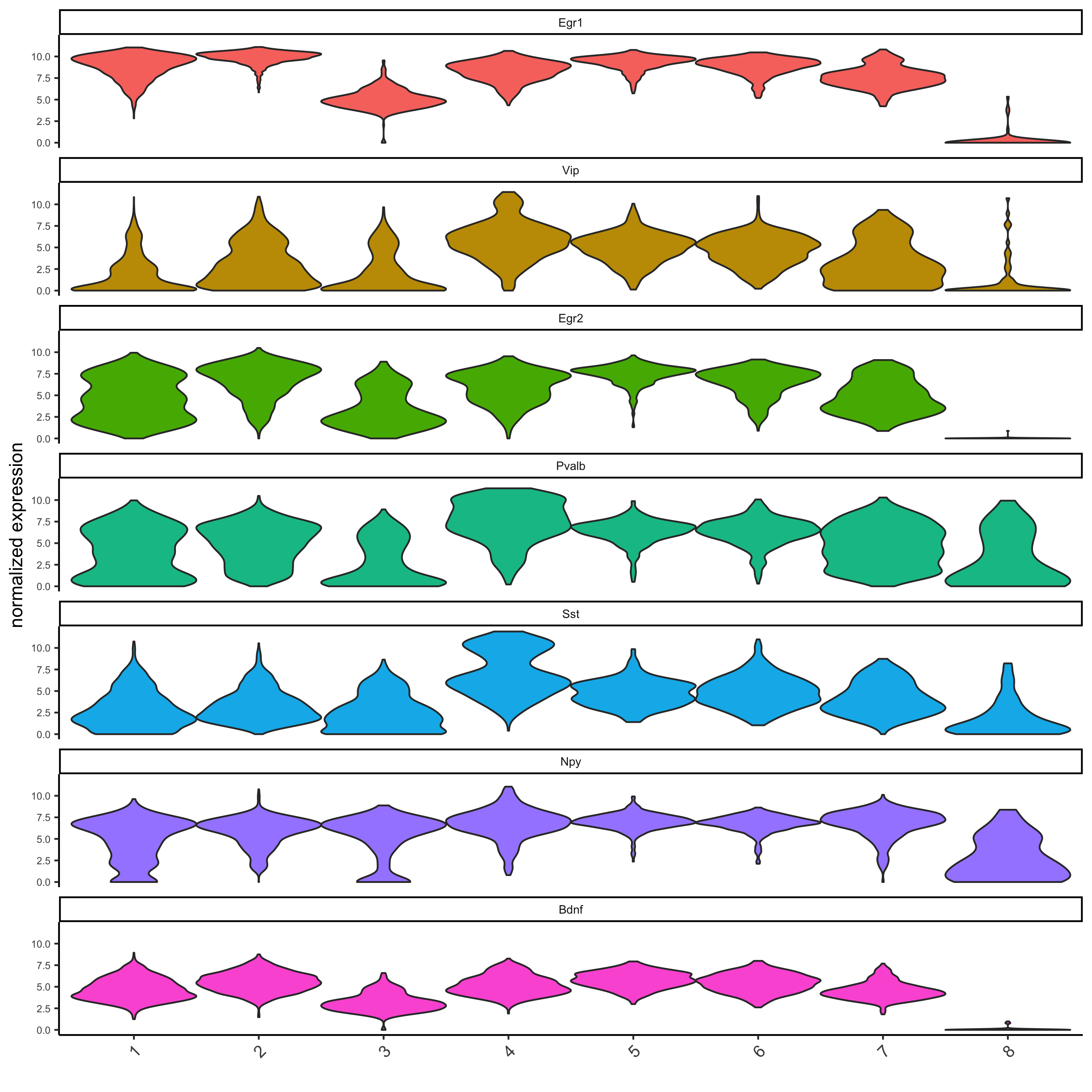
gini_markers = findMarkers_one_vs_all(gobject = starmap_mini, method = 'gini', expression_values = 'normalized', cluster_column = 'leiden_clus', min_genes = 20, min_expr_gini_score = 0.5, min_det_gini_score = 0.5) # get top 2 genes per cluster and visualize with violinplot topgenes_gini = gini_markers[, head(.SD, 2), by = 'cluster'] violinPlot(starmap_mini, genes = topgenes_gini$genes, cluster_column = 'leiden_clus', save_param = list(save_name = '5_a_violinplot'))
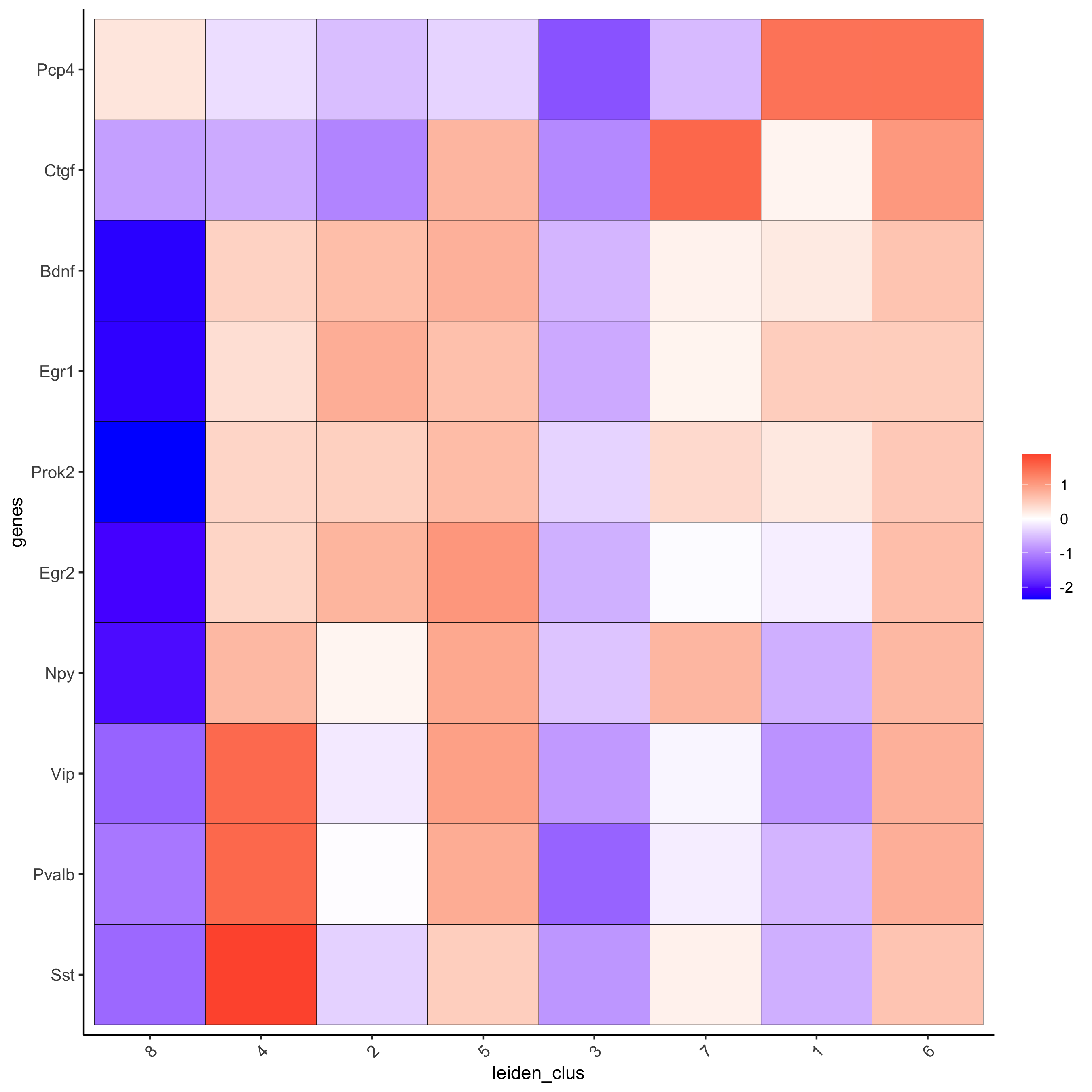
# get top 6 genes per cluster and visualize with heatmap topgenes_gini2 = gini_markers[, head(.SD, 6), by = 'cluster'] plotMetaDataHeatmap(starmap_mini, selected_genes = topgenes_gini2$genes, metadata_cols = c('leiden_clus'), save_param = list(save_name = '5_b_metaheatmap'))
6. A. cell type annotation
clusters_cell_types = c('cell A', 'cell B', 'cell C', 'cell D', 'cell E', 'cell F', 'cell G', 'cell H') names(clusters_cell_types) = 1:8 starmap_mini = annotateGiotto(gobject = starmap_mini, annotation_vector = clusters_cell_types, cluster_column = 'leiden_clus', name = 'cell_types') # check new cell metadata pDataDT(starmap_mini) # visualize annotations spatDimPlot(gobject = starmap_mini, cell_color = 'cell_types', spat_point_size = 2, dim_point_size = 2, save_param = list(save_name = '6_a_spatdimplot'))
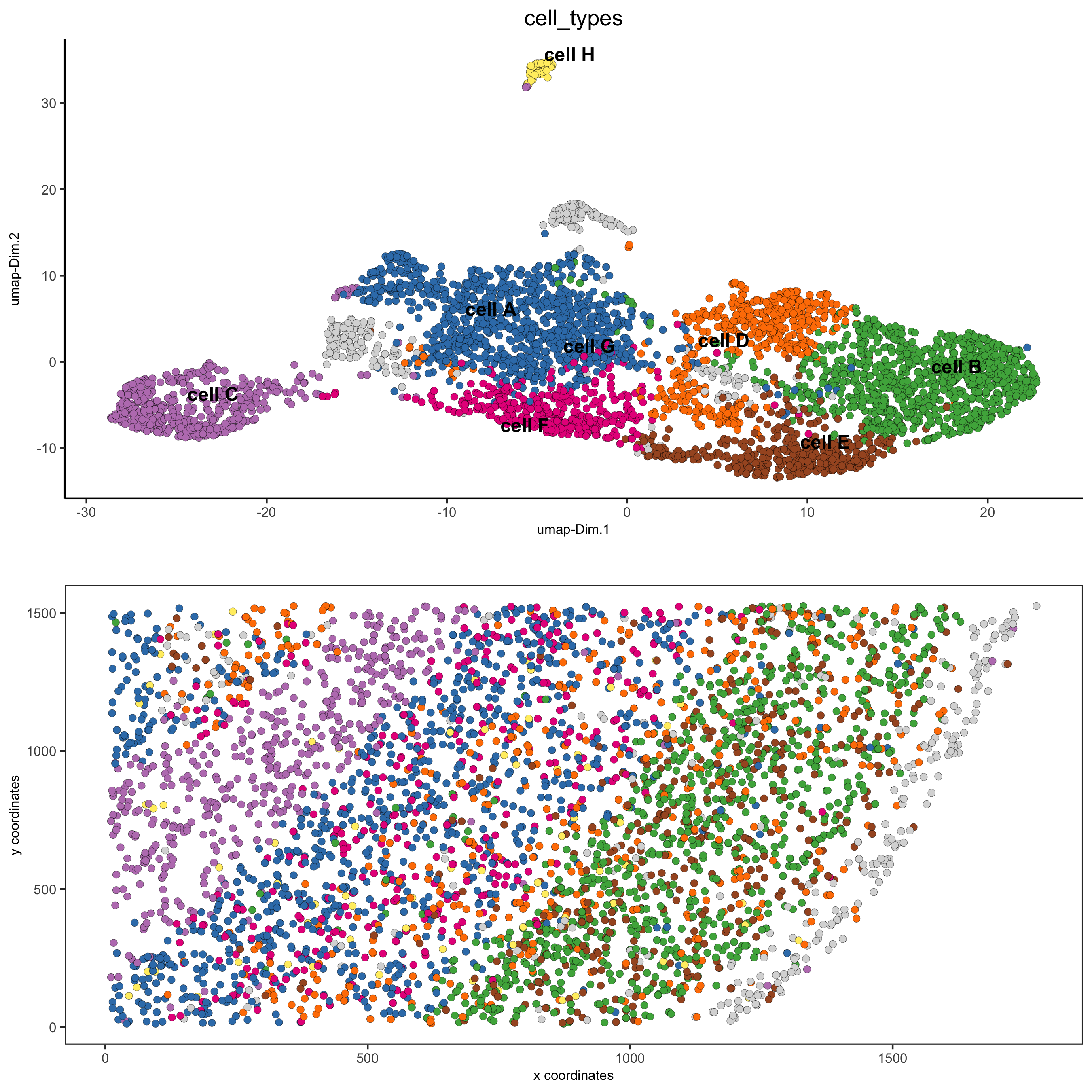
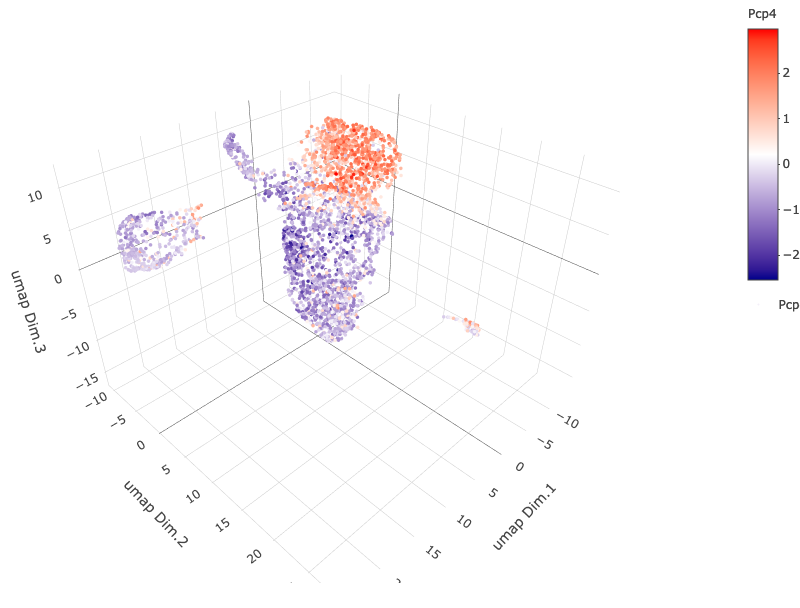
6. B. cell type gene expression
dimGenePlot3D(starmap_mini, dim_reduction_name = '3D_umap', expression_values = 'scaled', genes = "Pcp4", genes_high_color = 'red', genes_mid_color = 'white', genes_low_color = 'darkblue', save_param = list(save_name = '6_b_dimgeneplot'))
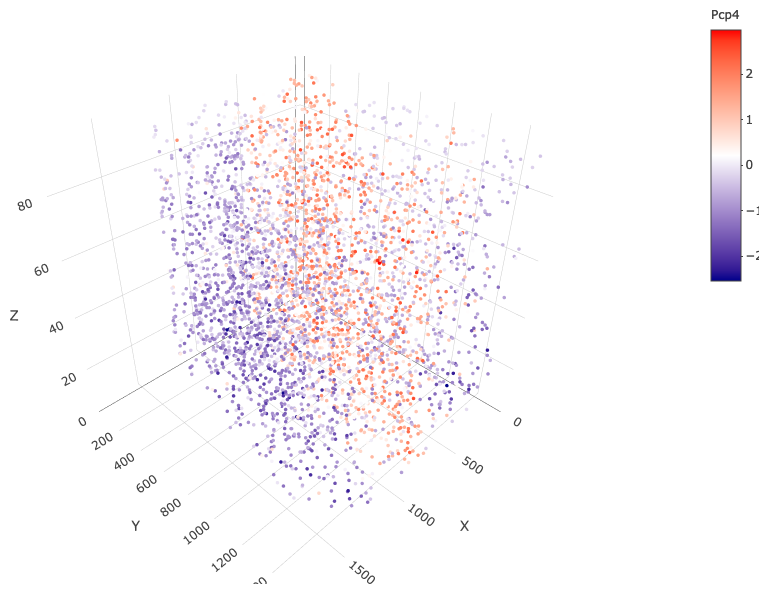
spatGenePlot3D(starmap_mini, expression_values = 'scaled', genes = "Pcp4", show_other_cells = F, genes_high_color = 'red', genes_mid_color = 'white', genes_low_color = 'darkblue', save_param = list(save_name = '6_c_spatgeneplot'))
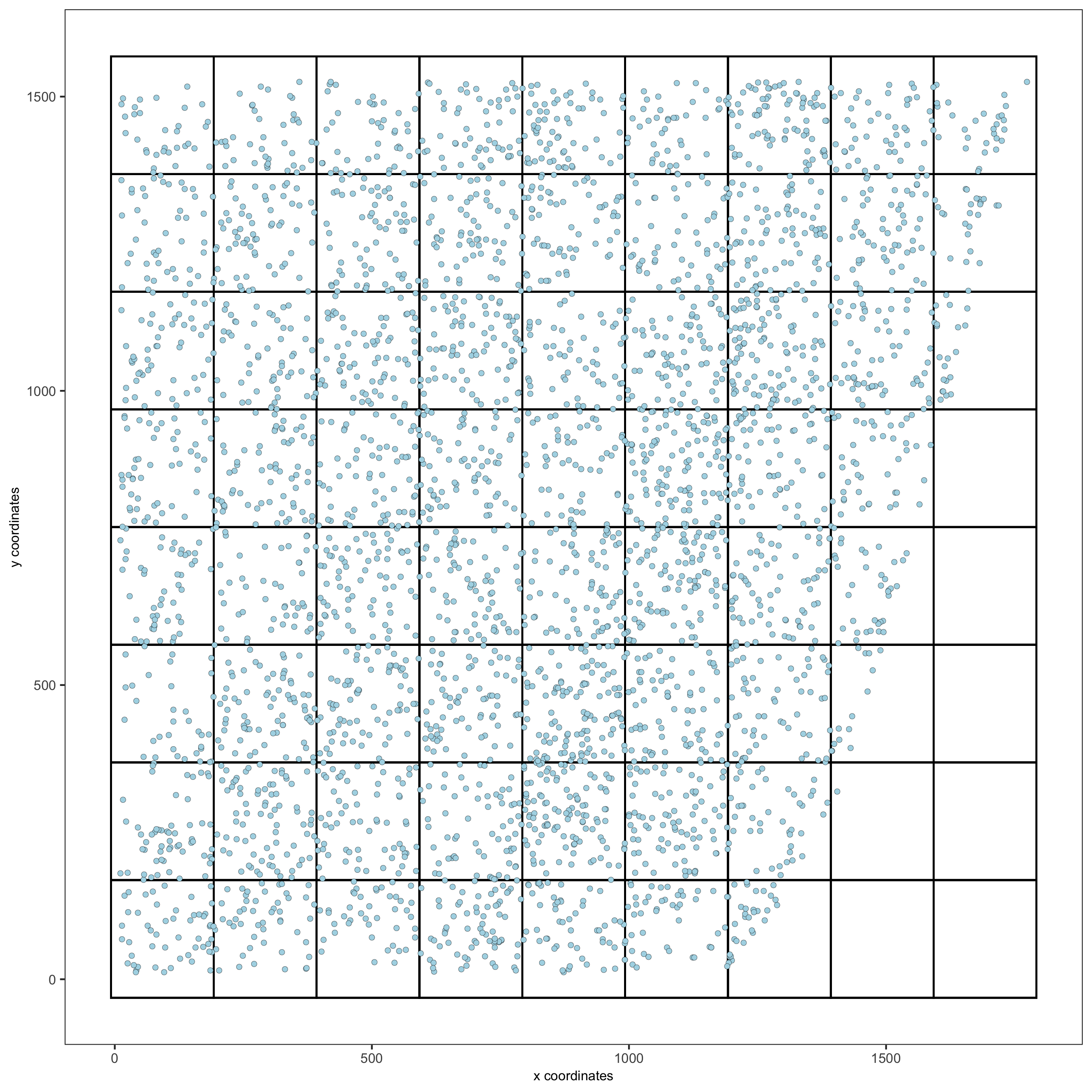
7. spatial grid
Create a grid based on defined stepsizes in the x,y(,z) axes.
starmap_mini <- createSpatialGrid(gobject = starmap_mini, sdimx_stepsize = 200, sdimy_stepsize = 200, sdimz_stepsize = 20, minimum_padding = 10) showGrids(starmap_mini) # visualize grid spatPlot2D(gobject = starmap_mini, show_grid = T, point_size = 1.5, save_param = list(save_name = '7_a_spatplot'))
8. spatial network
Only the method = delaunayn_geometry can make 3D Delaunay networks. This requires the package geometry to be installed.
- visualize information about the default Delaunay network
- create a spatial Delaunay network (default)
- create a spatial kNN network
plotStatDelaunayNetwork(gobject = starmap_mini, maximum_distance = 200, method = 'delaunayn_geometry', save_param = list(save_name = '8_aa_delnetwork'))
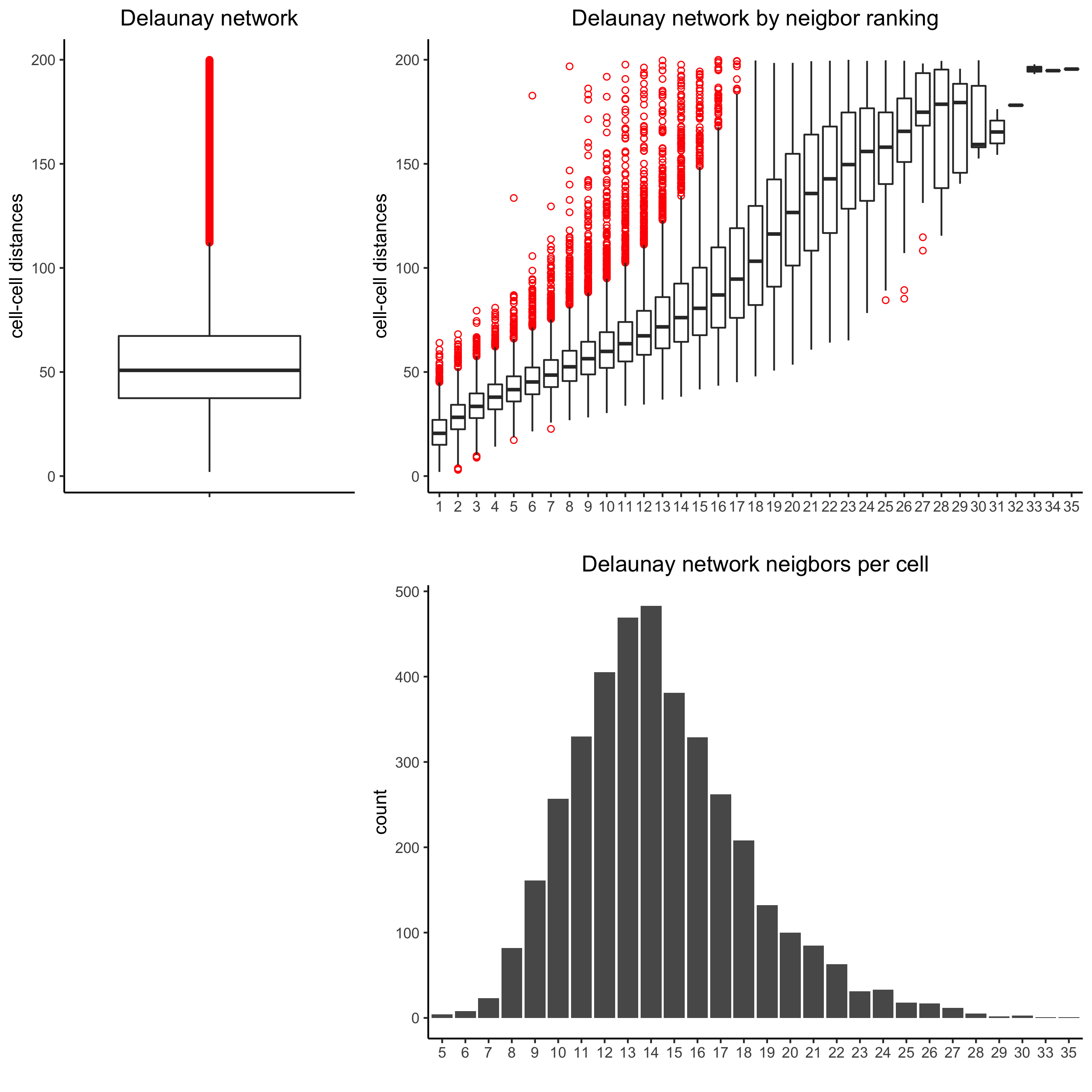
starmap_mini = createSpatialNetwork(gobject = starmap_mini, minimum_k = 2, maximum_distance_delaunay = 200, method = 'Delaunay', delaunay_method = 'delaunayn_geometry') starmap_mini = createSpatialNetwork(gobject = starmap_mini, minimum_k = 2, method = 'kNN', k = 10) showNetworks(starmap_mini) # visualize the two different spatial networks spatPlot(gobject = starmap_mini, show_network = T, network_color = 'blue', spatial_network_name = 'Delaunay_network', point_size = 2.5, cell_color = 'leiden_clus', save_param = list(save_name = '8_a_spatplot'))
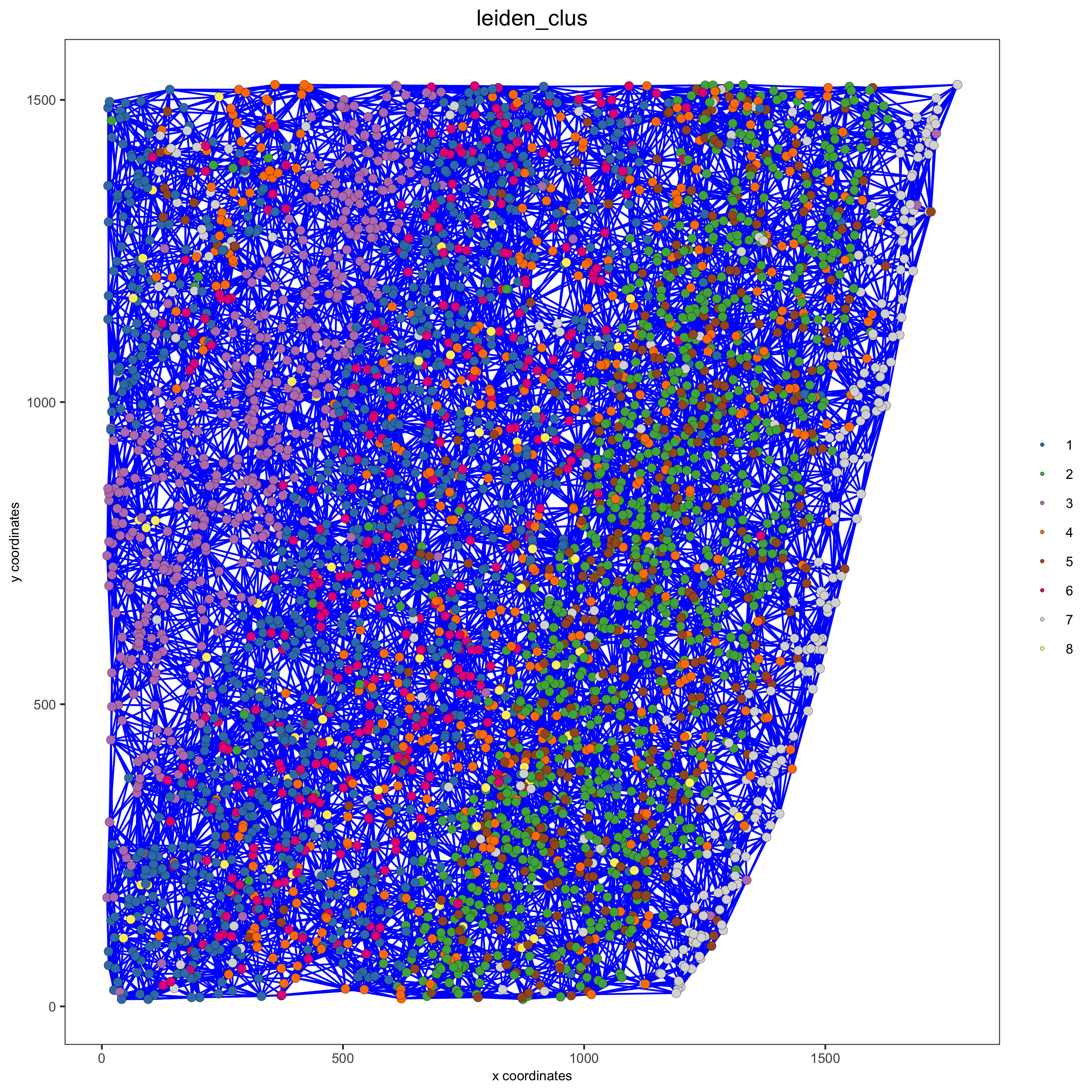
spatPlot(gobject = starmap_mini, show_network = T, network_color = 'blue', spatial_network_name = 'kNN_network', point_size = 2.5, cell_color = 'leiden_clus', save_param = list(save_name = '8_b_spatplot'))
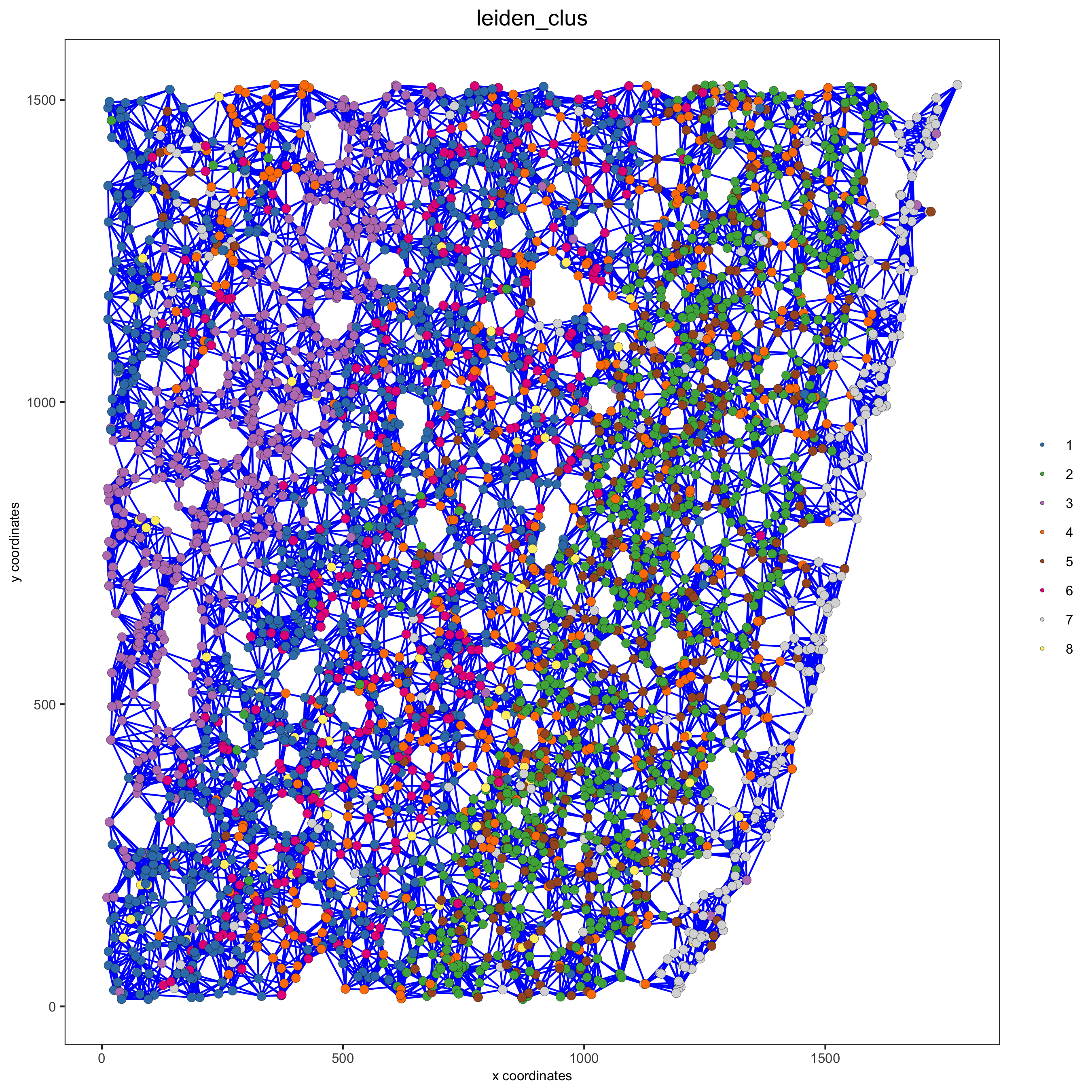
9. spatial genes
Identify spatial genes with 3 different methods:
- binSpect with kmeans binarization (default)
- binSpect with rank binarization
- silhouetteRank
Visualize top 4 genes per method.
km_spatialgenes = binSpect(starmap_mini) spatGenePlot(starmap_mini, expression_values = 'scaled', genes = km_spatialgenes[1:4]$genes, point_shape = 'border', point_border_stroke = 0.1, show_network = F, network_color = 'lightgrey', point_size = 2.5, cow_n_col = 2, save_param = list(save_name = '9_a_spatgeneplot'))
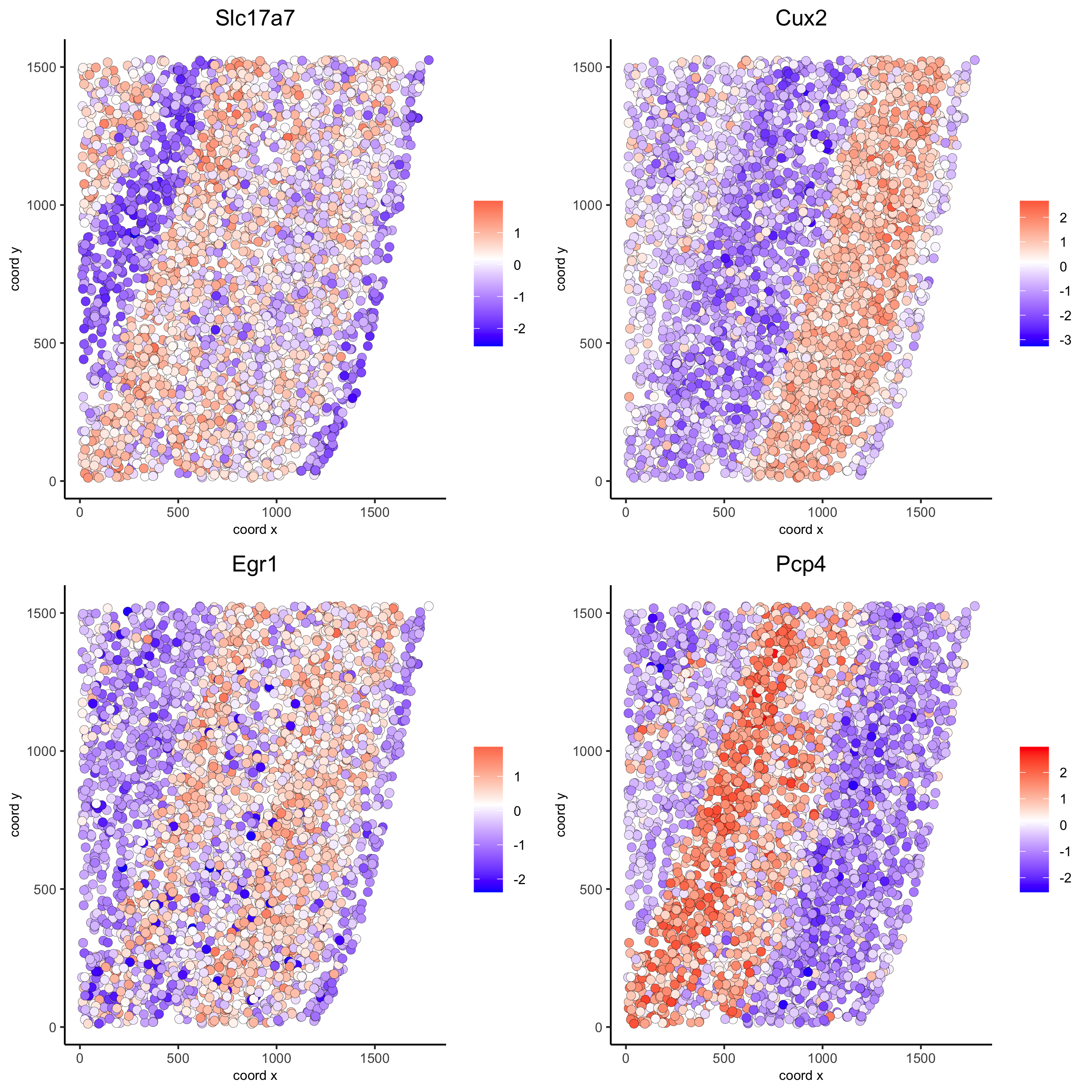
rank_spatialgenes = binSpect(starmap_mini, bin_method = 'rank') spatGenePlot(starmap_mini, expression_values = 'scaled', genes = rank_spatialgenes[1:4]$genes, point_shape = 'border', point_border_stroke = 0.1, show_network = F, network_color = 'lightgrey', point_size = 2.5, cow_n_col = 2, save_param = list(save_name = '9_b_spatgeneplot'))
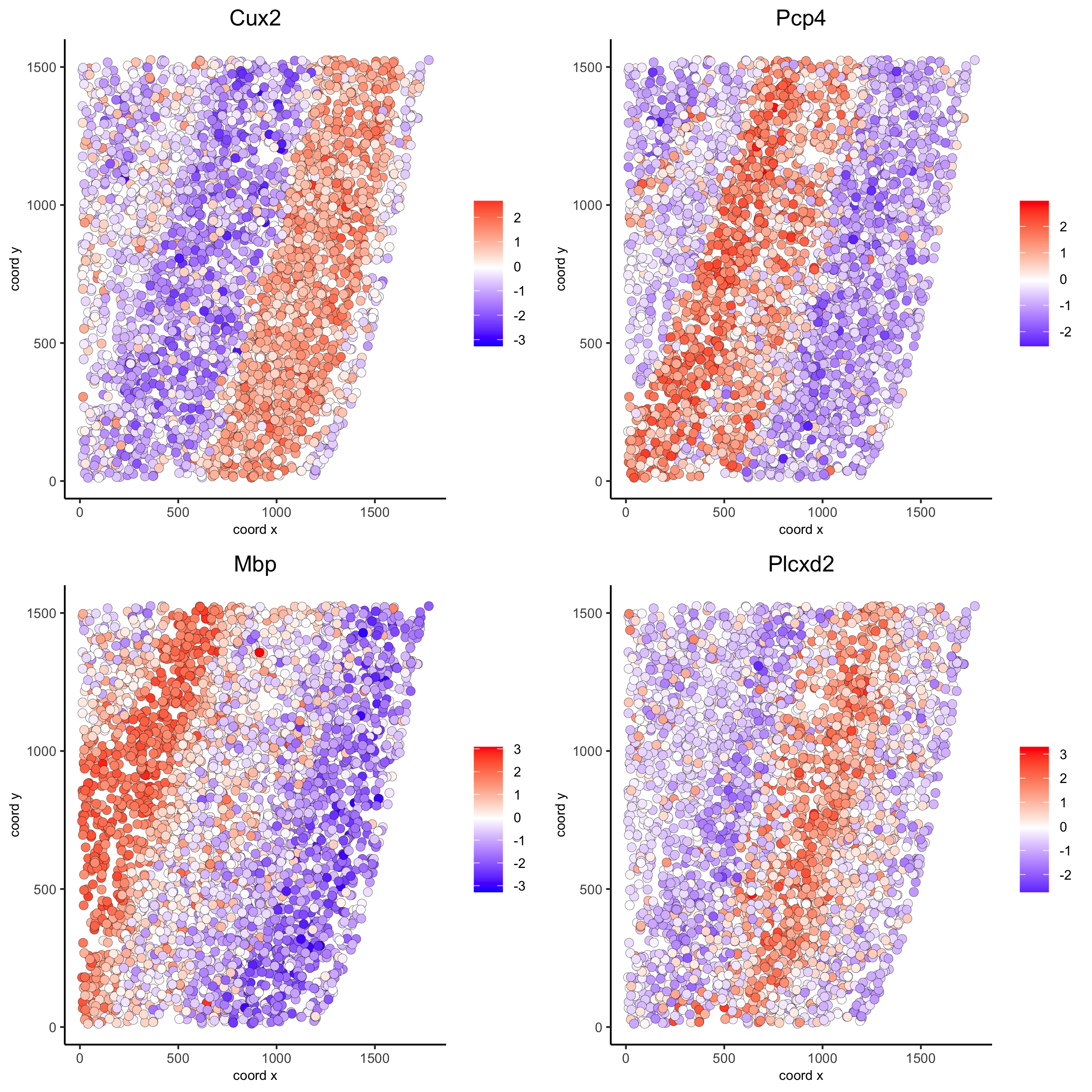
silh_spatialgenes = silhouetteRank(gobject = starmap_mini) # TODO: suppress print output spatGenePlot(starmap_mini, expression_values = 'scaled', genes = silh_spatialgenes[1:4]$genes, point_shape = 'border', point_border_stroke = 0.1, show_network = F, network_color = 'lightgrey', point_size = 2.5, cow_n_col = 2, save_param = list(save_name = '9_c_spatgeneplot'))
10. spatial co-expression patterns
Identify robust spatial co-expression patterns using the spatial network or grid and a subset of individual spatial genes.
1. calculate spatial correlation scores
2. cluster correlation scores
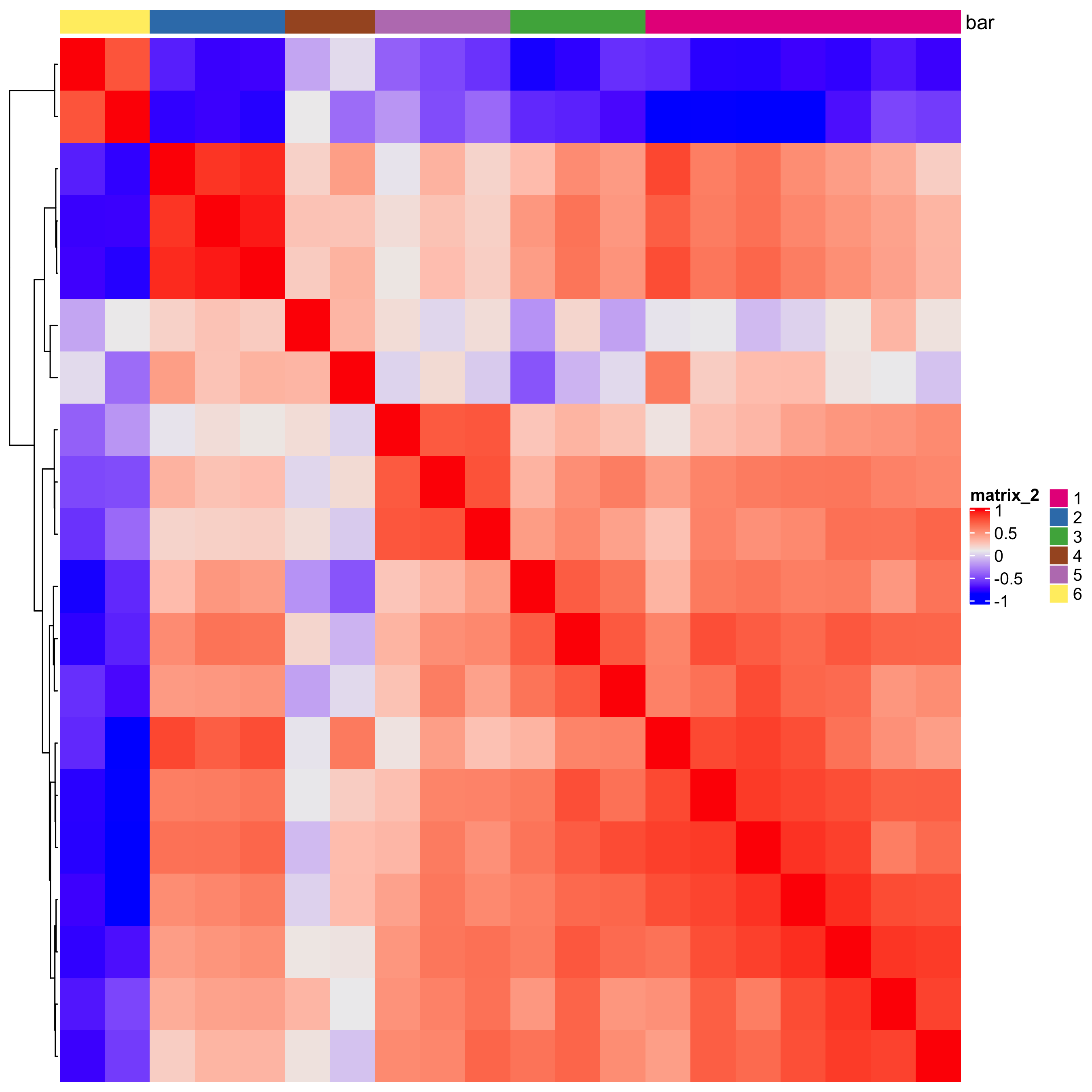
# 1. calculate spatial correlation scores ext_spatial_genes = km_spatialgenes[1:20]$genes spat_cor_netw_DT = detectSpatialCorGenes(starmap_mini, method = 'network', spatial_network_name = 'Delaunay_network', subset_genes = ext_spatial_genes) # 2. cluster correlation scores spat_cor_netw_DT = clusterSpatialCorGenes(spat_cor_netw_DT, name = 'spat_netw_clus', k = 6) heatmSpatialCorGenes(starmap_mini, spatCorObject = spat_cor_netw_DT, use_clus_name = 'spat_netw_clus', save_param = list(save_name = '10_a_heatmspatcor', units = 'in'))
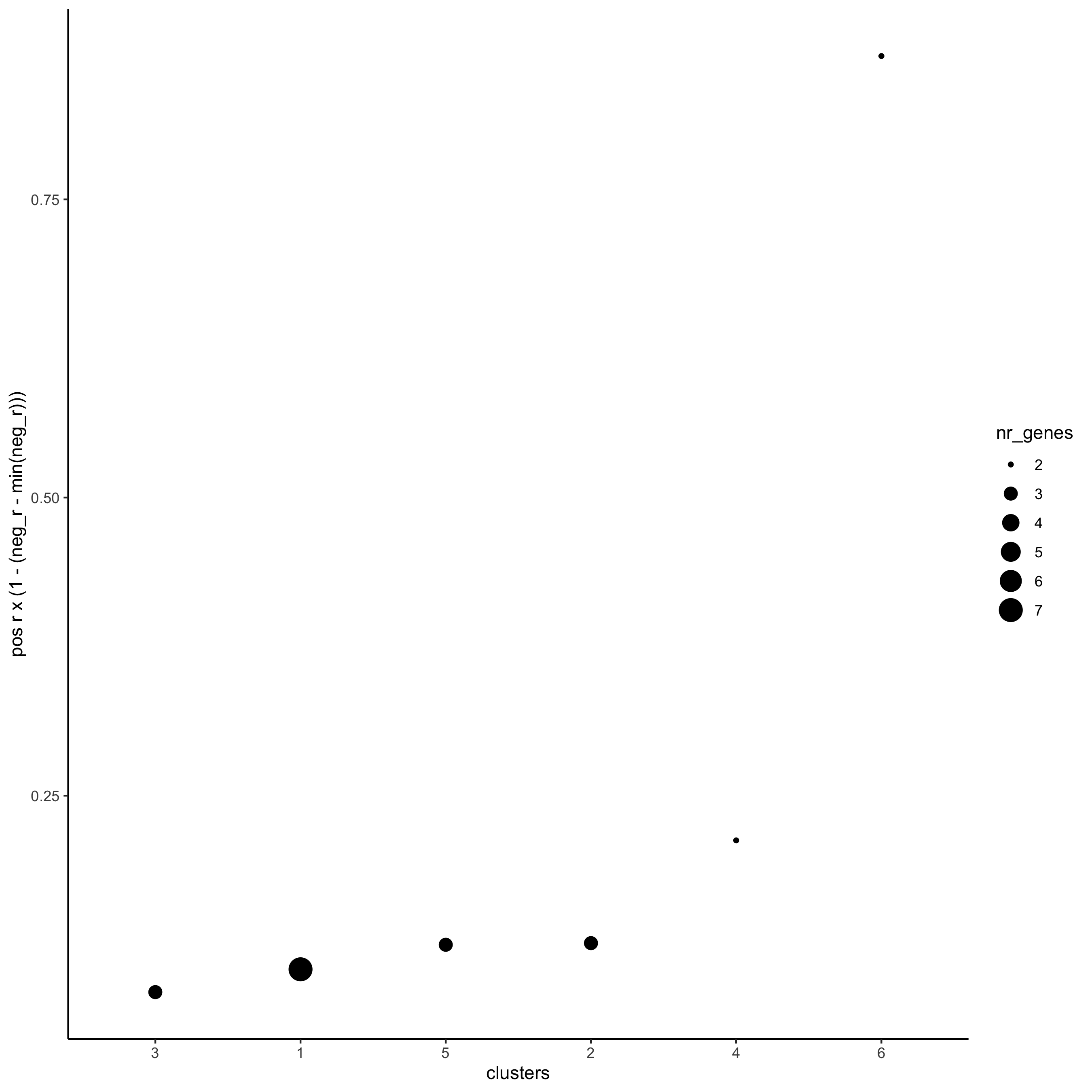
netw_ranks = rankSpatialCorGroups(starmap_mini, spatCorObject = spat_cor_netw_DT, use_clus_name = 'spat_netw_clus', save_param = list(save_name = '10_b_rankcorgroup'))
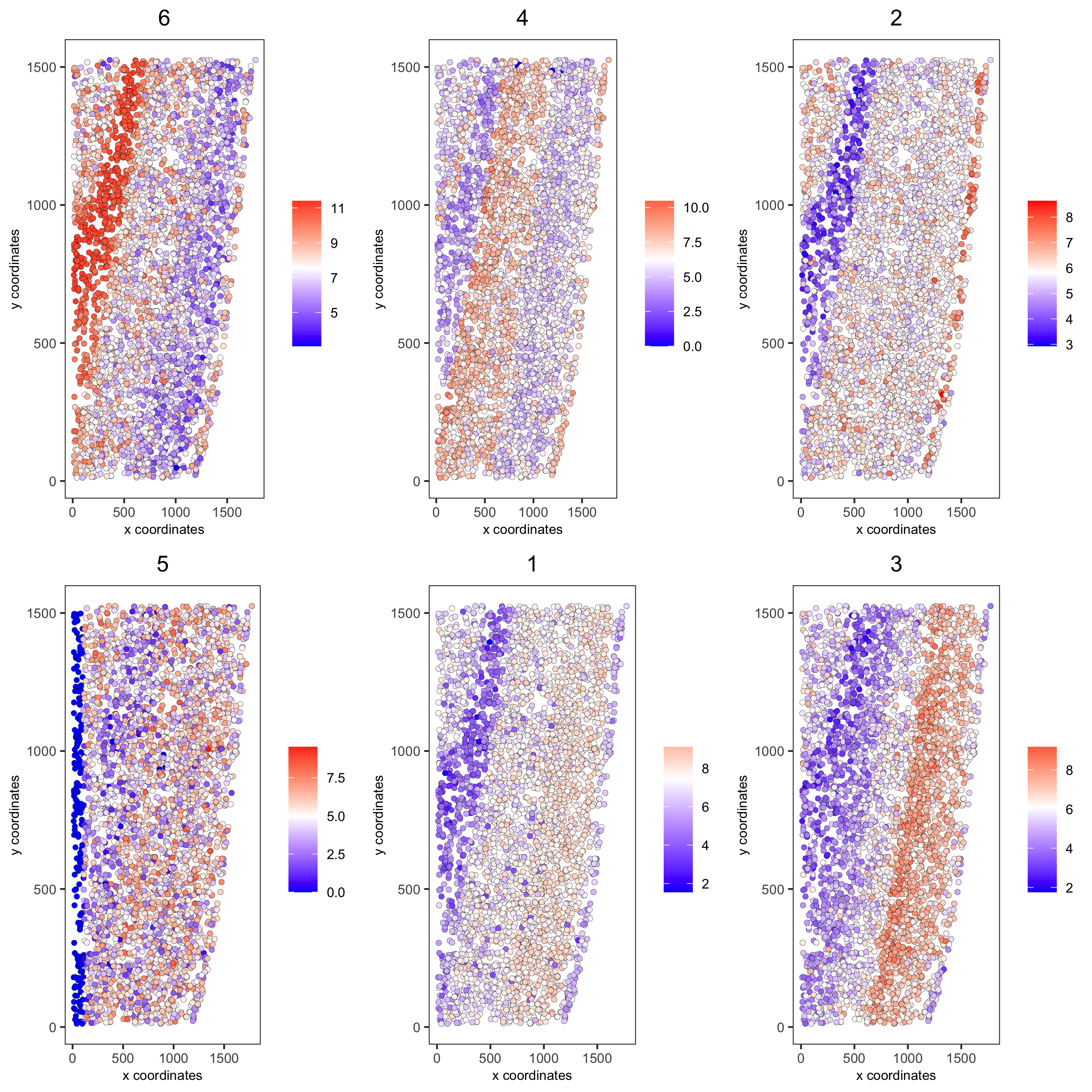
top_netw_spat_cluster = showSpatialCorGenes(spat_cor_netw_DT, use_clus_name = 'spat_netw_clus', selected_clusters = 6, show_top_genes = 1) cluster_genes_DT = showSpatialCorGenes(spat_cor_netw_DT, use_clus_name = 'spat_netw_clus', show_top_genes = 1) cluster_genes = cluster_genes_DT$clus; names(cluster_genes) = cluster_genes_DT$gene_ID starmap_mini = createMetagenes(starmap_mini, gene_clusters = cluster_genes, name = 'cluster_metagene') spatCellPlot(starmap_mini, spat_enr_names = 'cluster_metagene', cell_annotation_values = netw_ranks$clusters, point_size = 1.5, cow_n_col = 3, save_param = list(save_name = '10_c_spatcellplot'))
11. spatial HMRF domains
hmrf_folder = paste0(temp_dir,'/','11_HMRF/') if(!file.exists(hmrf_folder)) dir.create(hmrf_folder, recursive = T) # perform hmrf my_spatial_genes = km_spatialgenes[1:20]$genes HMRF_spatial_genes = doHMRF(gobject = starmap_mini, expression_values = 'scaled', spatial_genes = my_spatial_genes, spatial_network_name = 'Delaunay_network', k = 6, betas = c(10,2,2), output_folder = paste0(hmrf_folder, '/', 'Spatial_genes/SG_top20_k6_scaled')) # check and select hmrf for(i in seq(10, 14, by = 2)) { viewHMRFresults2D(gobject = starmap_mini, HMRFoutput = HMRF_spatial_genes, k = 6, betas_to_view = i, point_size = 2) } starmap_mini = addHMRF(gobject = starmap_mini, HMRFoutput = HMRF_spatial_genes, k = 6, betas_to_add = c(12), hmrf_name = 'HMRF') # visualize selected hmrf result giotto_colors = Giotto:::getDistinctColors(6) names(giotto_colors) = 1:6 spatPlot(gobject = starmap_mini, cell_color = 'HMRF_k6_b.12', point_size = 3, coord_fix_ratio = 1, cell_color_code = giotto_colors, save_param = list(save_name = '11_a_spatplot'))
12. cell neighborhood: cell-type/cell-type interactions
set.seed(seed = 2841) cell_proximities = cellProximityEnrichment(gobject = starmap_mini, cluster_column = 'cell_types', spatial_network_name = 'Delaunay_network', adjust_method = 'fdr', number_of_simulations = 1000) # barplot cellProximityBarplot(gobject = starmap_mini, CPscore = cell_proximities, min_orig_ints = 2, min_sim_ints = 2, p_val = 0.5, save_param = list(save_name = '12_a_barplot'))
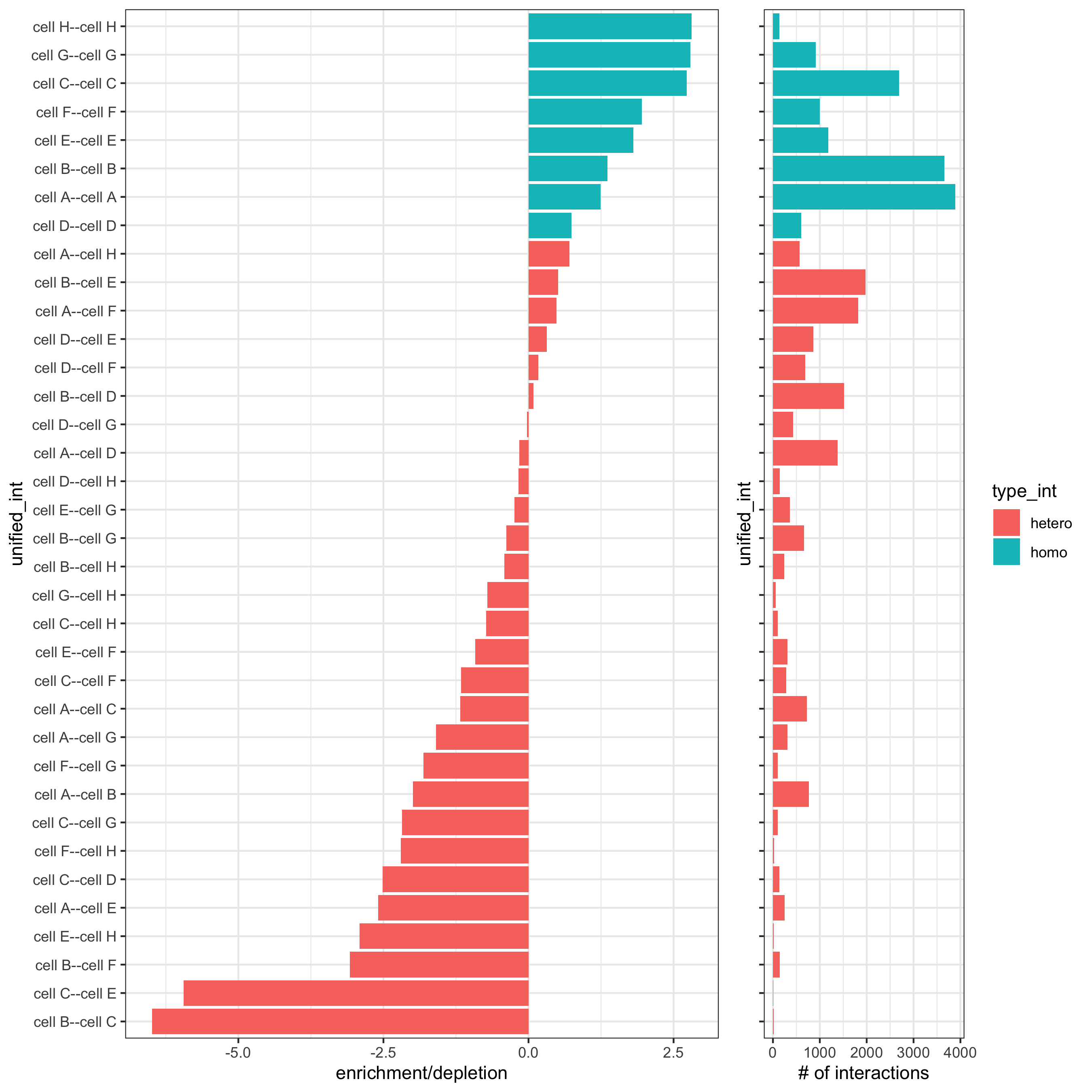
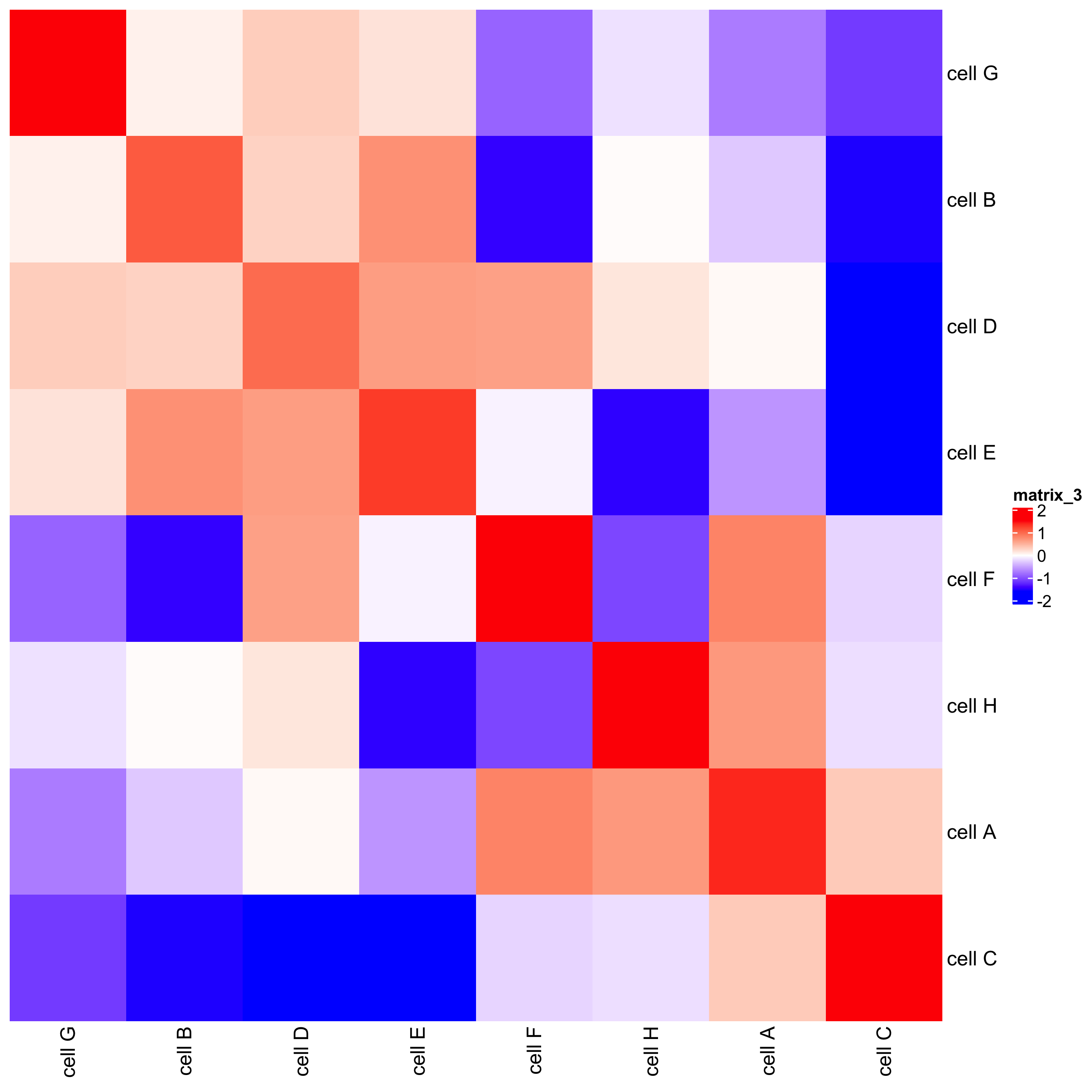
## heatmap cellProximityHeatmap(gobject = starmap_mini, CPscore = cell_proximities, order_cell_types = T, scale = T, color_breaks = c(-1.5, 0, 1.5), color_names = c('blue', 'white', 'red'), save_param = list(save_name = '12_b_heatmap', units = 'in'))
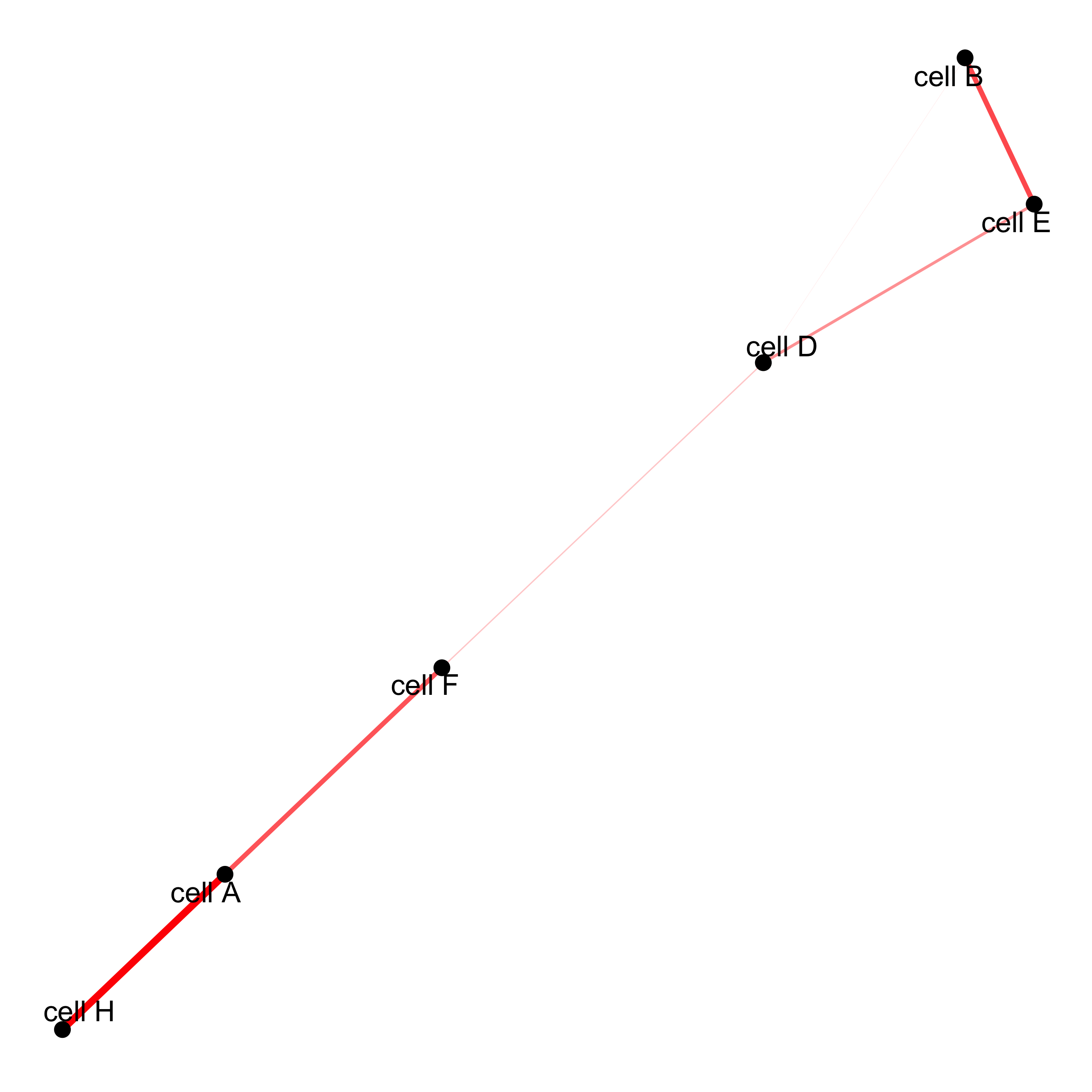
# network cellProximityNetwork(gobject = starmap_mini, CPscore = cell_proximities, remove_self_edges = T, only_show_enrichment_edges = T, save_param = list(save_name = '12_c_network'))
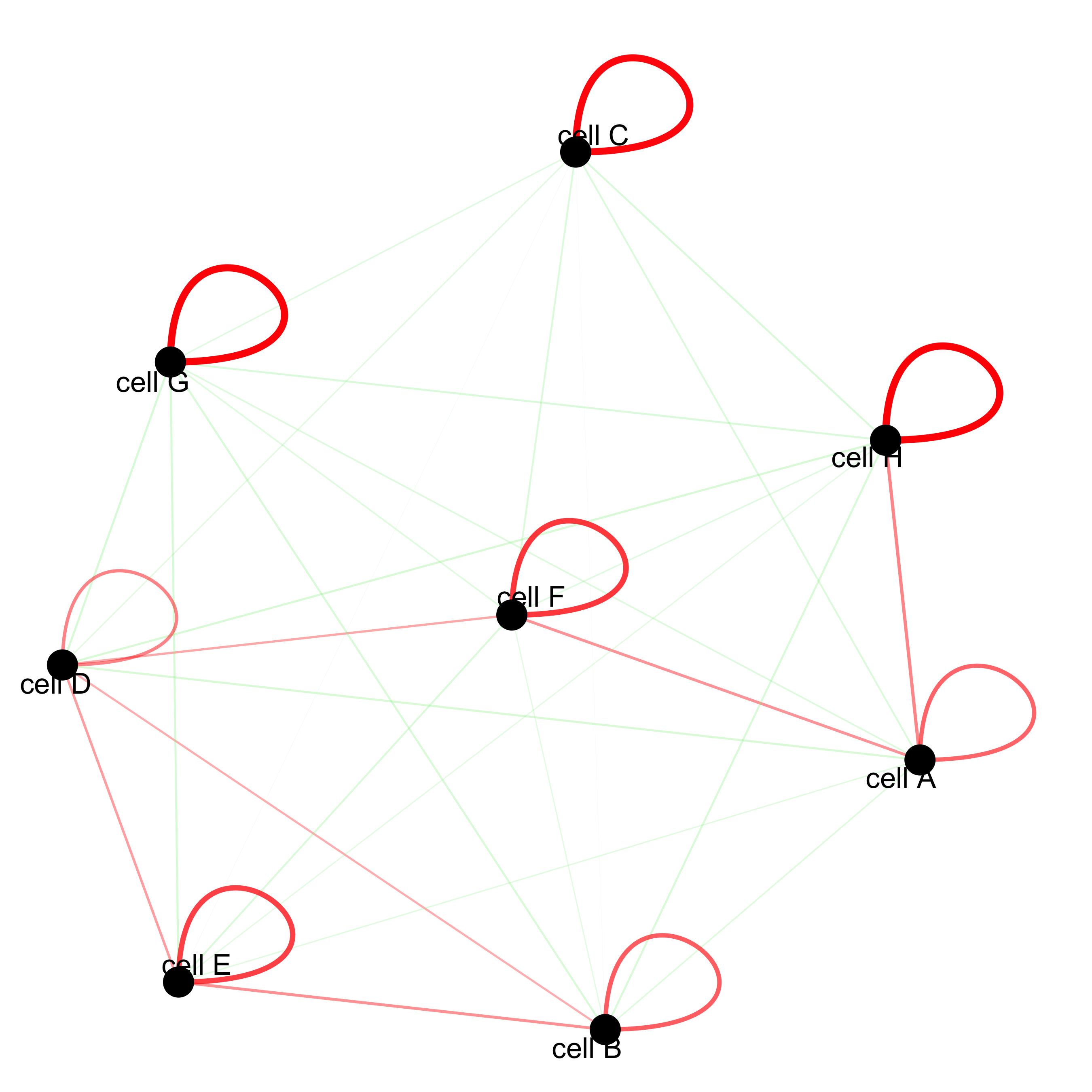
# network with self-edges cellProximityNetwork(gobject = starmap_mini, CPscore = cell_proximities, remove_self_edges = F, self_loop_strength = 0.3, only_show_enrichment_edges = F, rescale_edge_weights = T, node_size = 8, edge_weight_range_depletion = c(1, 2), edge_weight_range_enrichment = c(2,5), save_param = list(save_name = '12_d_network'))
visualization of specific cell types
pDataDT(starmap_mini) # Option 1 spec_interaction = "cell D--cell H" # needs to be in alphabetic order! first D, then H cellProximitySpatPlot2D(gobject = starmap_mini, interaction_name = spec_interaction, show_network = T, cluster_column = 'cell_types', cell_color = 'cell_types', cell_color_code = c('cell H' = 'lightblue', 'cell D' = 'red'), point_size_select = 4, point_size_other = 2, save_param = list(save_name = '12_e_cellproximity'))
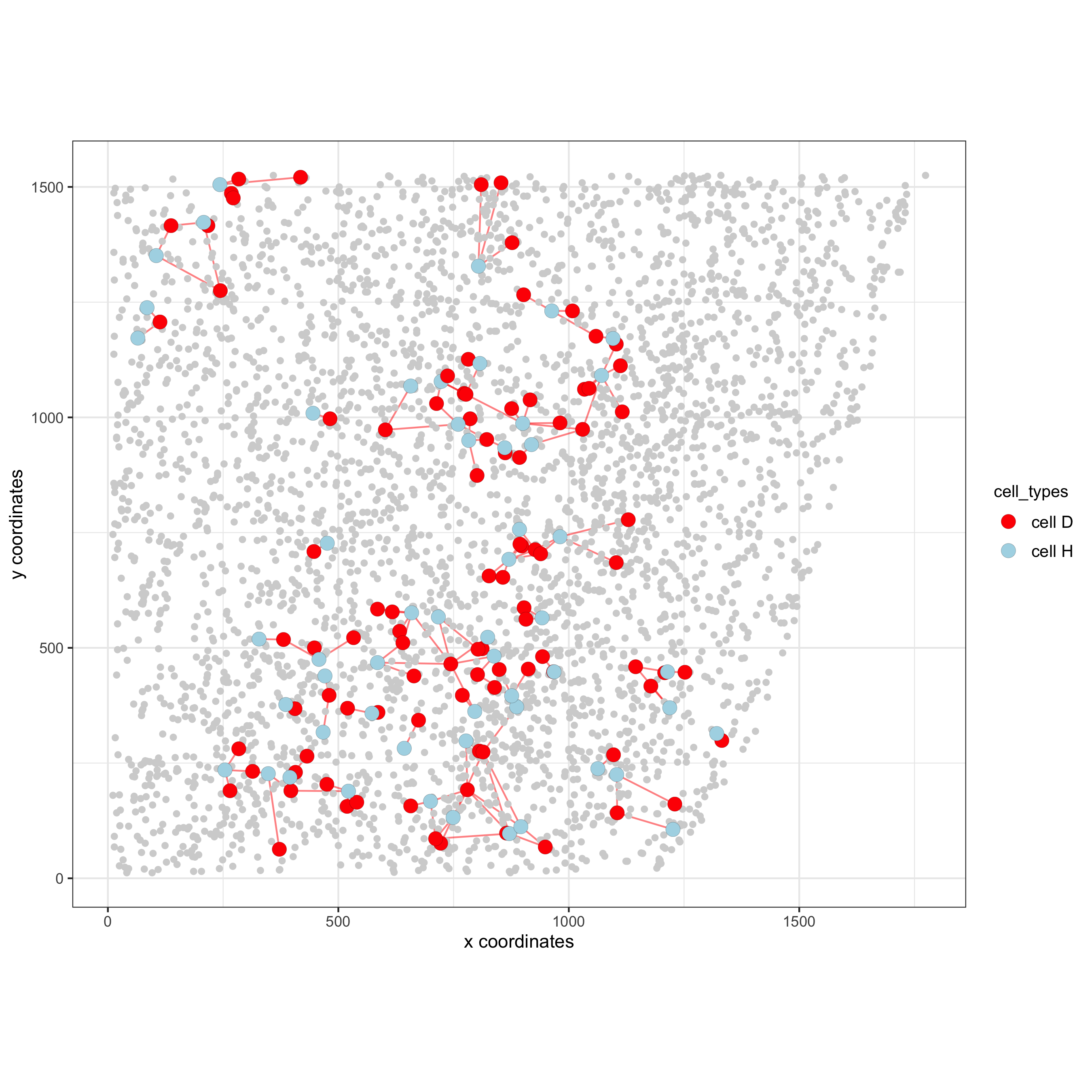
# Option 2: create additional metadata starmap_mini = addCellIntMetadata(starmap_mini, spatial_network = 'Delaunay_network', cluster_column = 'cell_types', cell_interaction = spec_interaction, name = 'D_H_interactions') spatPlot(starmap_mini, cell_color = 'D_H_interactions', legend_symbol_size = 3, select_cell_groups = c('other_cell D', 'other_cell H', 'select_cell D', 'select_cell H'), save_param = list(save_name = '12_e_spatplot'))
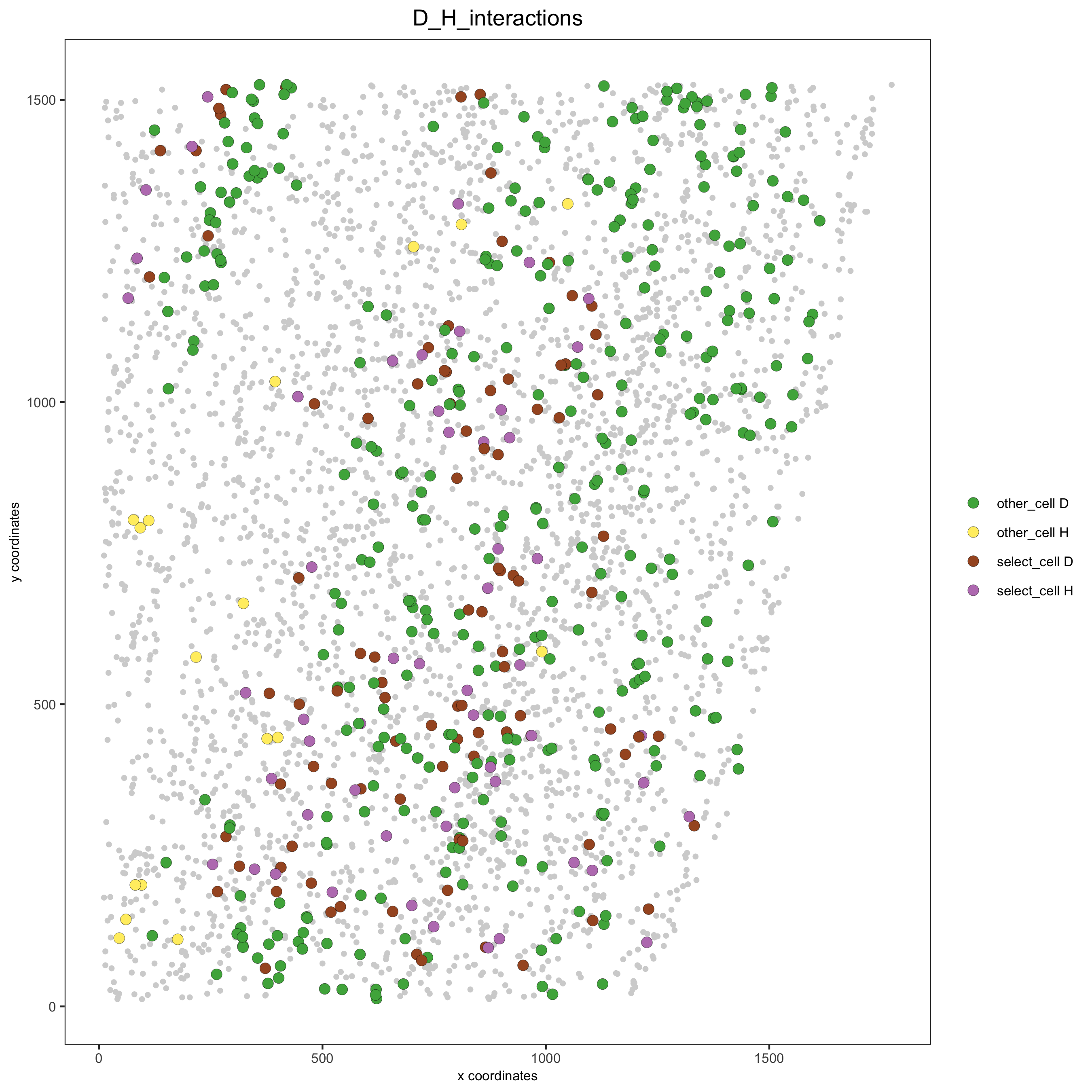
13. 2D cross sections from 3D object
# create cross section starmap_mini = createCrossSection(starmap_mini, method="equation", equation=c(0,1,0,600), extend_ratio = 0.6) # show cross section insertCrossSectionSpatPlot3D(starmap_mini, cell_color = 'leiden_clus', axis_scale = 'cube', point_size = 2, save_param = list(save_name = '13_a_insertcross'))
insertCrossSectionGenePlot3D(starmap_mini, expression_values = 'scaled', axis_scale = "cube", genes = "Slc17a7", save_param = list(save_name = '13_b_insertcrossgene'))
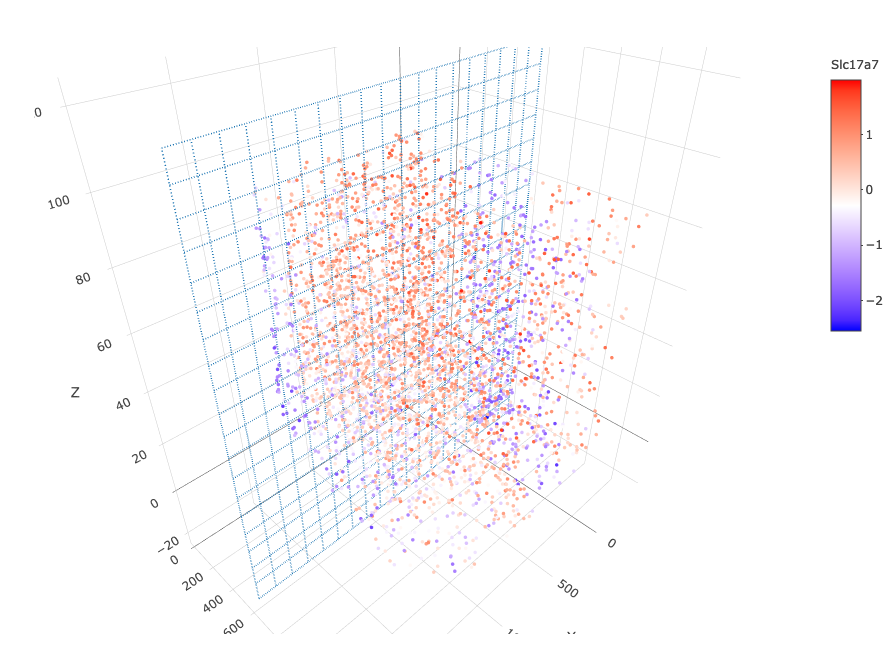
# for cell annotation crossSectionPlot(starmap_mini, point_size = 2, point_shape = "border", cell_color = "leiden_clus", save_param = list(save_name = '13_c_crossplot'))
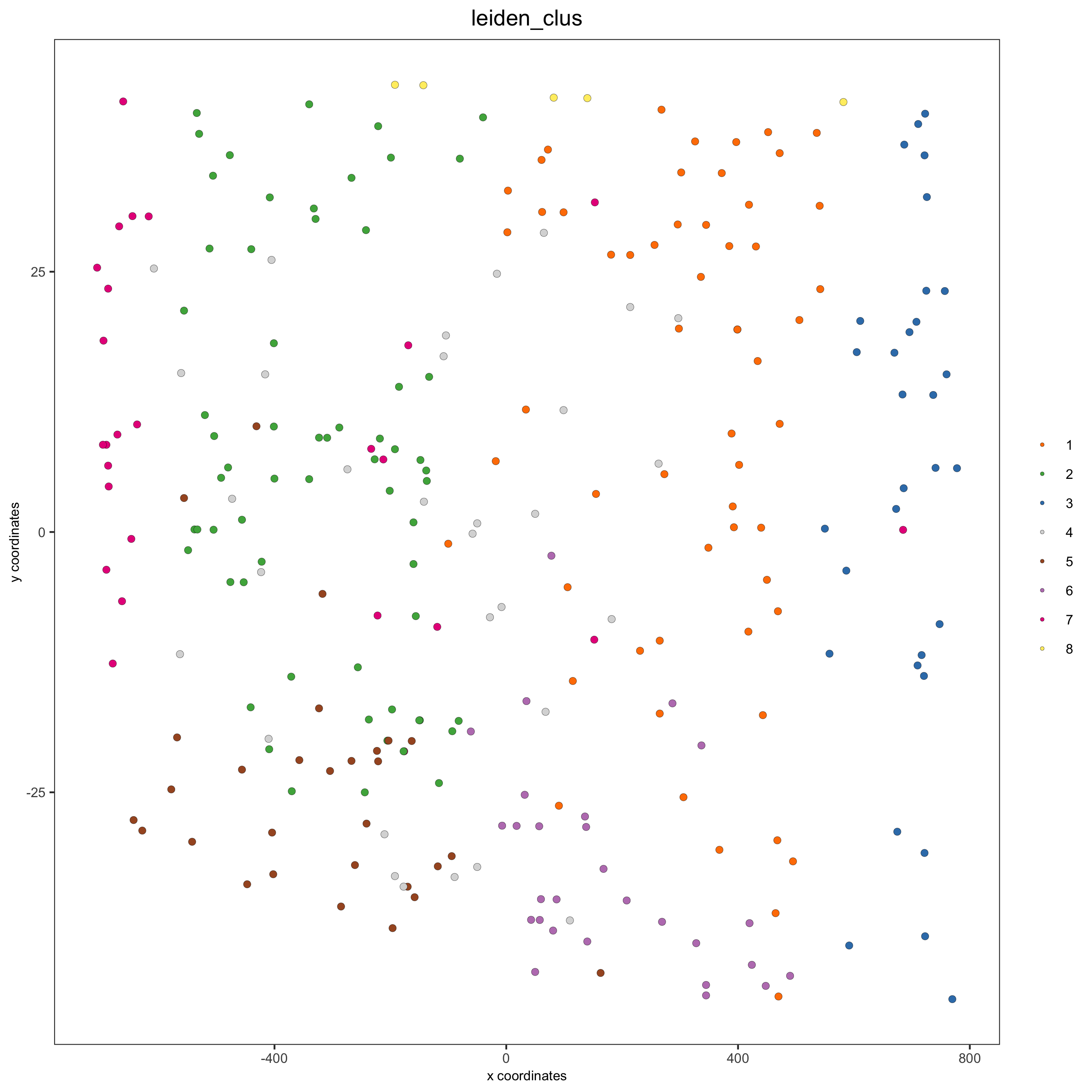
crossSectionPlot3D(starmap_mini, point_size = 2, cell_color = "leiden_clus", axis_scale = "cube", save_param = list(save_name = '13_c_crossplot3D'))
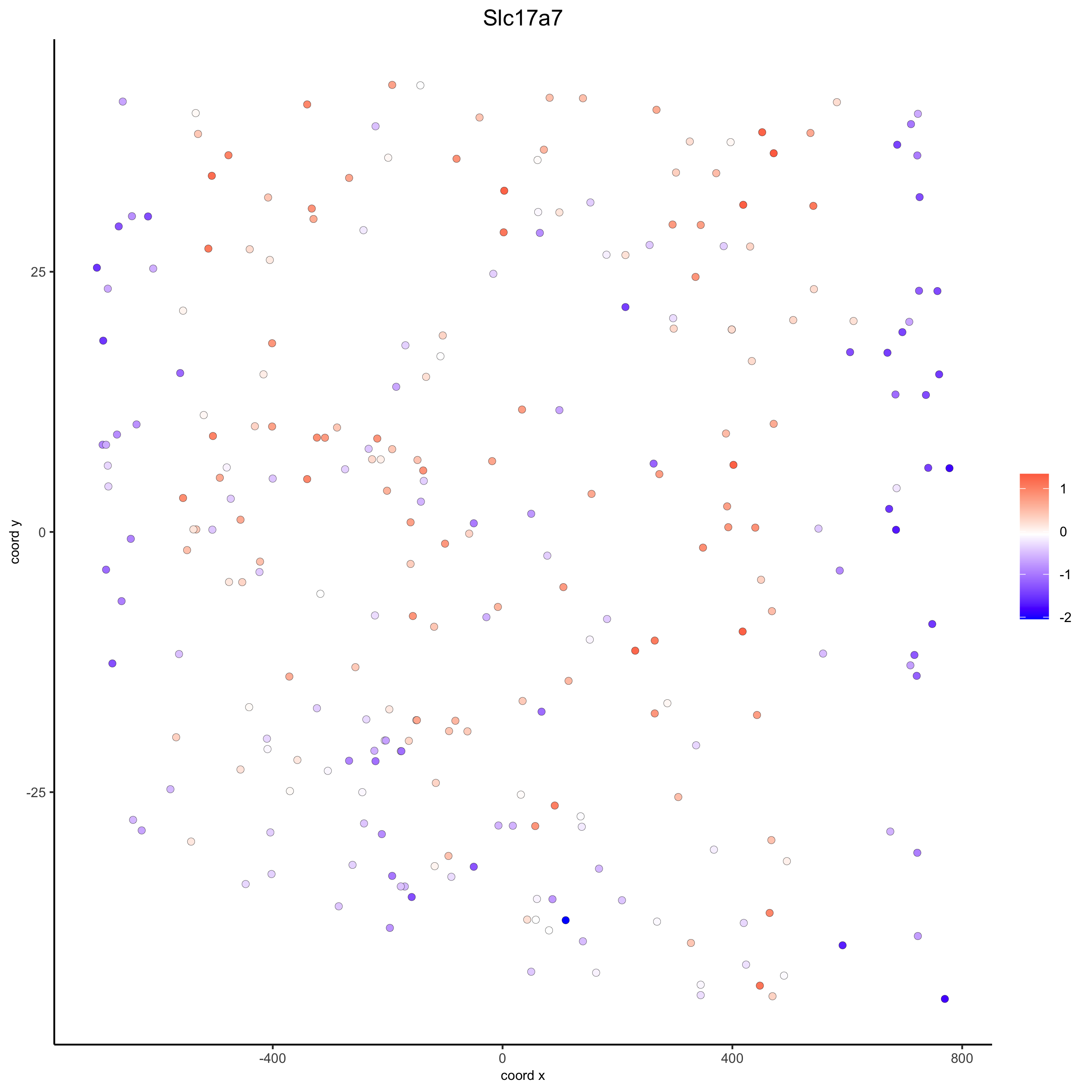
# for gene expression crossSectionGenePlot(starmap_mini, genes = "Slc17a7", point_size = 2, point_shape = "border", cow_n_col = 1.5, expression_values = 'scaled', save_param = list(save_name = '13_d_crossgeneplot'))
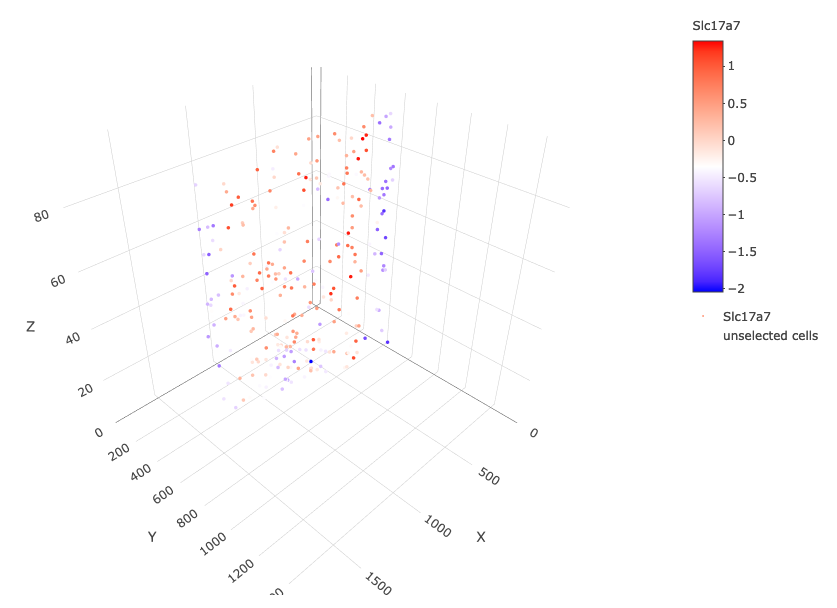
crossSectionGenePlot3D(starmap_mini, point_size = 2, genes = c("Slc17a7"), expression_values = 'scaled', save_param = list(save_name = '13_e_crossgeneplot3D'))
14. export Giotto Analyzer to Viewer
viewer_folder = paste0(temp_dir, '/', 'Mouse_cortex_viewer') # select annotations, reductions and expression values to view in Giotto Viewer exportGiottoViewer(gobject = starmap_mini, output_directory = viewer_folder, factor_annotations = c('cell_types', 'leiden_clus', 'HMRF_k6_b.12'), numeric_annotations = 'total_expr', dim_reductions = c('umap'), dim_reduction_names = c('umap'), expression_values = 'scaled', expression_rounding = 3, overwrite_dir = T)